Integration of a Large-Scale Genetic Analysis Workbench Increases the Accessibility of a High-Performance Pathway-Based Analysis Method
Article information

Abstract
The rapid increase in genetic dataset volume has demanded extensive adoption of biological knowledge to reduce the computational complexity, and the biological pathway is one well-known source of such knowledge. In this regard, we have introduced a novel statistical method that enables the pathway-based association study of large-scale genetic dataset—namely, PHARAOH. However, researcher-level application of the PHARAOH method has been limited by a lack of generally used file formats and the absence of various quality control options that are essential to practical analysis. In order to overcome these limitations, we introduce our integration of the PHARAOH method into our recently developed all-in-one workbench. The proposed new PHARAOH program not only supports various de facto standard genetic data formats but also provides many quality control measures and filters based on those measures. We expect that our updated PHARAOH provides advanced accessibility of the pathway-level analysis of large-scale genetic datasets to researchers.
Introduction
Recently, many genetic association studies have sought larger sample sizes for various reasons, such as the rapid decline of sequencing costs and small effect sizes [1]. The computational complexity required to analyze such datasets has in turn become larger; hence, many researchers have developed novel association analysis methods by adopting additional information to reasonably reduce the dimension of datasets [2, 3]. In this respect, we have proposed the PHARAOH method, which is a computationally efficient method that can analyze large-scale genetic datasets—for example, thousands of samples with millions of variants [4]. By applying doubly penalized regression to the hierarchical model that mimics the underlying biological structure from genes to the phenotype via pathways, we have demonstrated that the proposed method can handle substantial correlations between pathways but analyzes large-scale datasets within a reasonable time with superior statistical power.
However, it is also important that a widely used method is easy to use, in addition to its methodological advantages. In that regard, the PHARAOH program lacks accessibility to researchers, based on limited support of file formats and the absence of integrated quality control processes and computational flexibility. To address this problem, we introduce an updated PHARAOH program that is integrated with our recently published all-in-one genetic workbench, WISARD [5]. WISARD is a powerful workbench that accepts a broad range of input formats and provides extensive quality control measures that can be calculated and used to filter out samples or variants, before the various analyses that WISARD supports. By using the advantages of WISARD, we integrated three features of WISARD into the PHARAOH program: input format support, quality control, and multithreading.
Implementation
An overall workflow of the updated PHARAOH program is depicted in Fig. 1. Following the implementation of the WISARD framework, the program retrieves genotypes from a given dataset and additional files (phenotype, covariates, gene and pathway mapping) through the WISARD framework. The program now accepts four mainstream genotype formats (PLINK PED, transposed PED, long-format GEN, and Variant Call Format [VCF]), through integration of the WISARD framework.
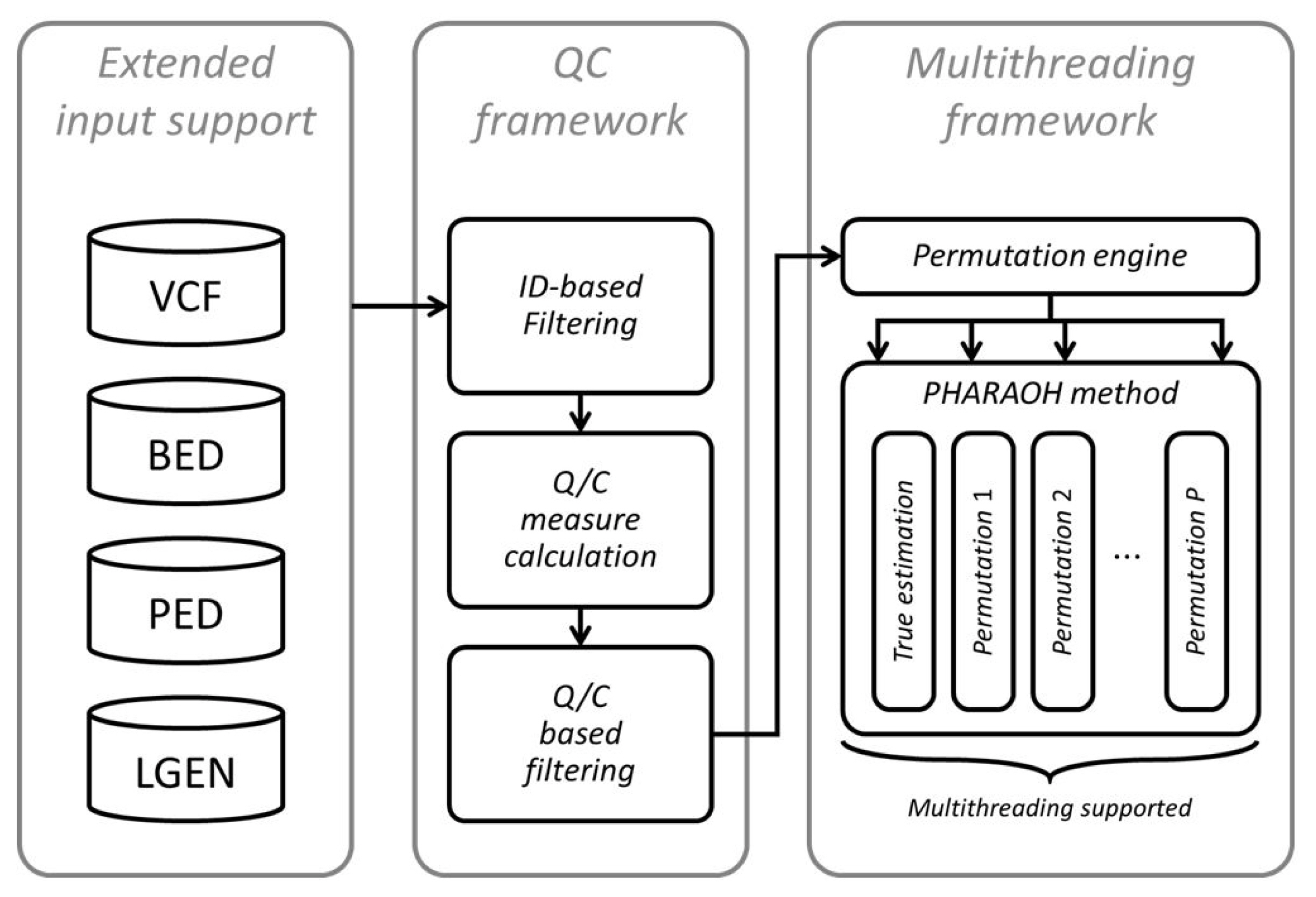
Workflow of the new PHARAOH program. VCF, Variant Calling Format.
Next, the program computes given quality control measures and then filters samples and variants based on the criteria. In this step, more than 30 options can be applied to generate various quality control measures and filters based on those measures. Details on the measures can be found at the WISARD website (http://statgen.snu.ac.kr/wisard/). Moreover, we also implemented PHARAOH-specific quality control measures in addition to the WISARD measures, for filtering of unqualified genes or pathways.
After fixation of the dataset, the analysis can be done in a multithreaded way. Multithreading was applied to two parts of the PHARAOH algorithm. We applied the multithreading scheme of WISARD to the (1) automated estimation step of penalization parameters and (2) parameter estimation from permuted phenotypes to construct an empirical null distribution, because a series of parameter combinations or a model with permuted phenotypes can be evaluated independently. As shown in Table 1, which shows the result of PHARAOH multithreading, the multithreaded PHARAOH analysis can be boosted as the number of threads increases.

Comparison of execution times by number of threads. All results were averaged from the same three runs
Conclusion
In this paper, we introduced recent progress with our previously developed method. While there were several advantages of the proposed method, limited accessibility of the implementation of the program has hindered the utilization of the PHARAOH method so far. However, we successfully showed that a combination of a well-developed workbench that covers the limitation of the method can improve its accessibility to researchers, through our integration of the PHARAOH method and the WISARD framework.
Acknowledgments
This work was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI16C2037).
Notes
Availability: An updated version of PHARAOH is available at http://statgen.snu.ac.kr/software/pharaoh/.
Authors’ contribution
Conceptualization: SL
Funding acquisition: TP
Methodology: SL, TP
Writing – original draft: SL
Writing – review & editing: SL, TP
Conflicts of Interest
No potential conflicts of interest relevant to this article was reported.