CTCF, Cohesin, and Chromatin in Human Cancer
Article information

Abstract
It is becoming increasingly clear that eukaryotic genomes are subjected to higher-order chromatin organization by the CCCTC-binding factor/cohesin complex. Their dynamic interactions in three dimensions within the nucleus regulate gene transcription by changing the chromatin architecture. Such spatial genomic organization is functionally important for the spatial disposition of chromosomes to control cell fate during development and differentiation. Thus, the dysregulation of proper long-range chromatin interactions may influence the development of tumorigenesis and cancer progression.
Introduction
Genomes form higher-order chromatin structures during cellular development and differentiation [1–4]. The spatial folding of chromosomes and their organization in the nucleus have profound effects on gene expression [5, 6]. It is now well established that chromosomal architecture is largely mediated by the CCCTC-binding factor (CTCF)/cohesin complex [7, 8]. CTCF, a zinc finger DNA-binding protein that functions in transcriptional repression, activation, and as an insulator that interferes with enhancer–promoter interactions [9], is needed for the recruitment of cohesin to chromatin [10]. In addition to its major influence on sister chromatid cohesion [11], cohesin affects gene transcription by facilitating long-range interactions between members of many developmentally regulated gene families [12–14].
Interphase chromosomes of higher eukaryotes are subdivided into evolutionarily conserved topologically associated domains (TADs) in the three-dimensional space of the nucleus (Fig. 1) [15, 16]. TADs are defined as self-associating chromosome segments enclosed by a chromatin loop in the megabase range detected by Hi-C methods [17]. TADs show a high frequency of interactions within domains and a low frequency of interactions among different domains [15, 17]. They are partitioned into several subcompartments related to their gene expression patterns and maintained across cell types, suggesting that TADs shape the regulatory landscape of the genome during development [6, 15, 16]. Interestingly, in the boundaries of TADs, there is an abundance of the architectural protein CTCF and cohesin [6, 15], suggesting that these proteins have a role in establishing the topological boundaries [17].
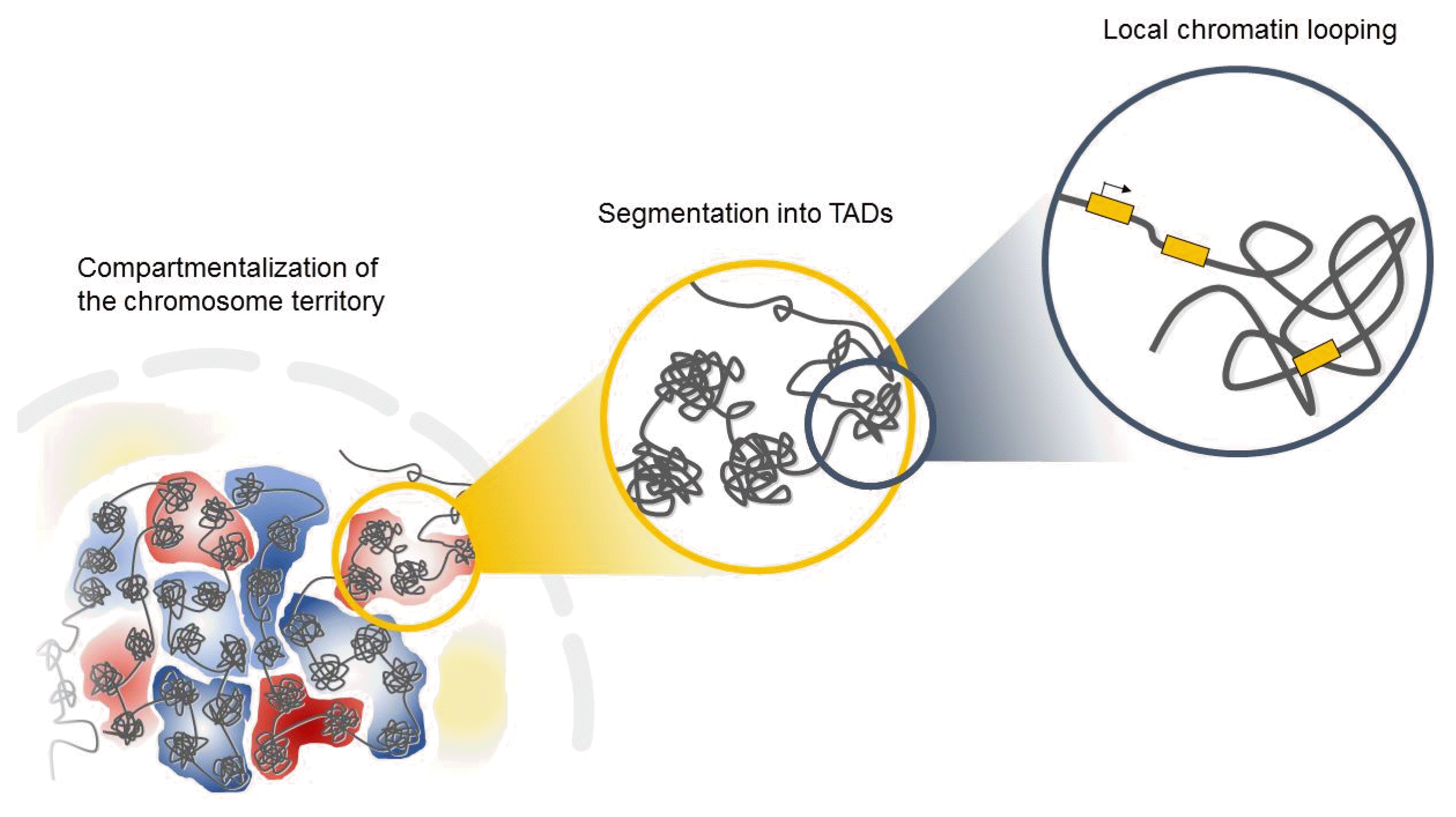
The spatial folding of chromosomes in the nucleus. TADs, topologically associated domains.
Increasing evidence from recent studies has indicated that genomic instability is spatially related to higher-order chromatin organization in cancer cells [18]. Previous studies of c-Myc, BCL, and immunoglobulin heavy-chain (Igh) (the most common translocation partners in various B-cell lymphomas) showed that these loci are preferentially localized in close spatial juxtaposition to each other in normal B cells [19, 20]. Moreover, the boundaries of copy-number alterations are preferentially localized in close spatial juxtaposition within the nucleus [21–23]. Thus, the continuous DNA damage and subsequent defective repair near double-strand break (DSB) sites might generate copy-number alterations of oncogenes during tumorigenesis [24, 25]. In addition, the recent discovery that DSBs and subsequent amplification of estrogen response elements can be generated by estrogen-induced long-range chromatin interactions in breast cancer [26] further indicates the importance of higher-order genome architecture for chromosomal rearrangement. Therefore, the close spatial proximity of genomic regions may provide an opportunity for the formation of specific, cancer-related chromosomal translocation events during tumor development [18, 27, 28].
Here, we briefly review recent works on the roles of CTCF and cohesin in genome folding, their global impact on gene expression, and their association with human disease.
Results
CTCF
Chromatin organizer roles of CTCF
CTCF is a sequence-specific DNA-binding protein that functions by utilizing an 11-zinc-finger domain [29]. Because CTCF was first identified at the 5′ and 3′ ends of the chicken β-globin locus [30] and the imprinted Igf2/H19 locus [31, 32], it is known as an insulator protein that can block enhancer activity in eukaryotes [32]. Although insulators would be expected to be located in intergenic regions where they could act as barriers to block enhancer activity [30], genome-wide analysis indicated that CTCF-binding sites are present in genes and/or promoter regions as well as intergenic regions [10, 33]. More recent evidence revealed that almost 15% of CTCF-recognition sites are located near promoters and ~40% are within exons and introns [17], suggesting that CTCF has dynamic roles other than enhancer blocking activity.
While earlier studies implied that the distribution patterns of CTCF are similar to those of transcription activators or repressors, recently determined global distribution patterns suggested that CTCF-binding sites are not strongly correlated with general transcription factor occupancy [10]. Moreover, depletion of CTCF altered its histone acetylation and methylation profiles in the β-globin locus, but did not significantly affect β-globin expression [34, 35], suggesting that CTCF has a role distinct from that of traditional regulatory proteins.
Interestingly, CTCF has been shown to serve as a chromatin organizer complex by linking chromosomal domains in the mouse/human β-globin cluster (Fig. 2) [36, 37]. During erythroid differentiation, CTCF is recruited and enables enhancers to physically access promoters of β-globin, which both influences transcription and contributes to cell-type-specific chromatin organization and function [36, 37]. Similarly, long-range interactions associated with CTCF have been observed within mammalian gene loci including the Igf2/H19 imprinted control region [38, 39], the α-globin gene cluster in erythroid cells [40], and the Igh locus in B cells [41].

A schematic representation of CCCTC-binding factor (CTCF)–mediated looping structure. TFs, transcription factors.
DNA methylation and CTCF binding
It has been known for many years that CTCF binding is abolished by the DNA methylation of CpG sites within the CTCF motif [32]. At the imprinted Igf2/H19 locus, CTCF binds specifically to the unmethylated differentially methylated region (DMR), which is required for the expression of H19 on the maternal chromosome (Fig. 3A) [32, 42]. However, on the paternal allele, the methylated DMR prohibits CTCF enrichment and leads to IGF2 expression [30, 42], suggesting methylation-sensitive binding of CTCF at the target region. Interestingly, genome-wide association studies have identified that only a small subset of CTCF-binding sites are sensitive to the methylation status of DNA [8, 43].
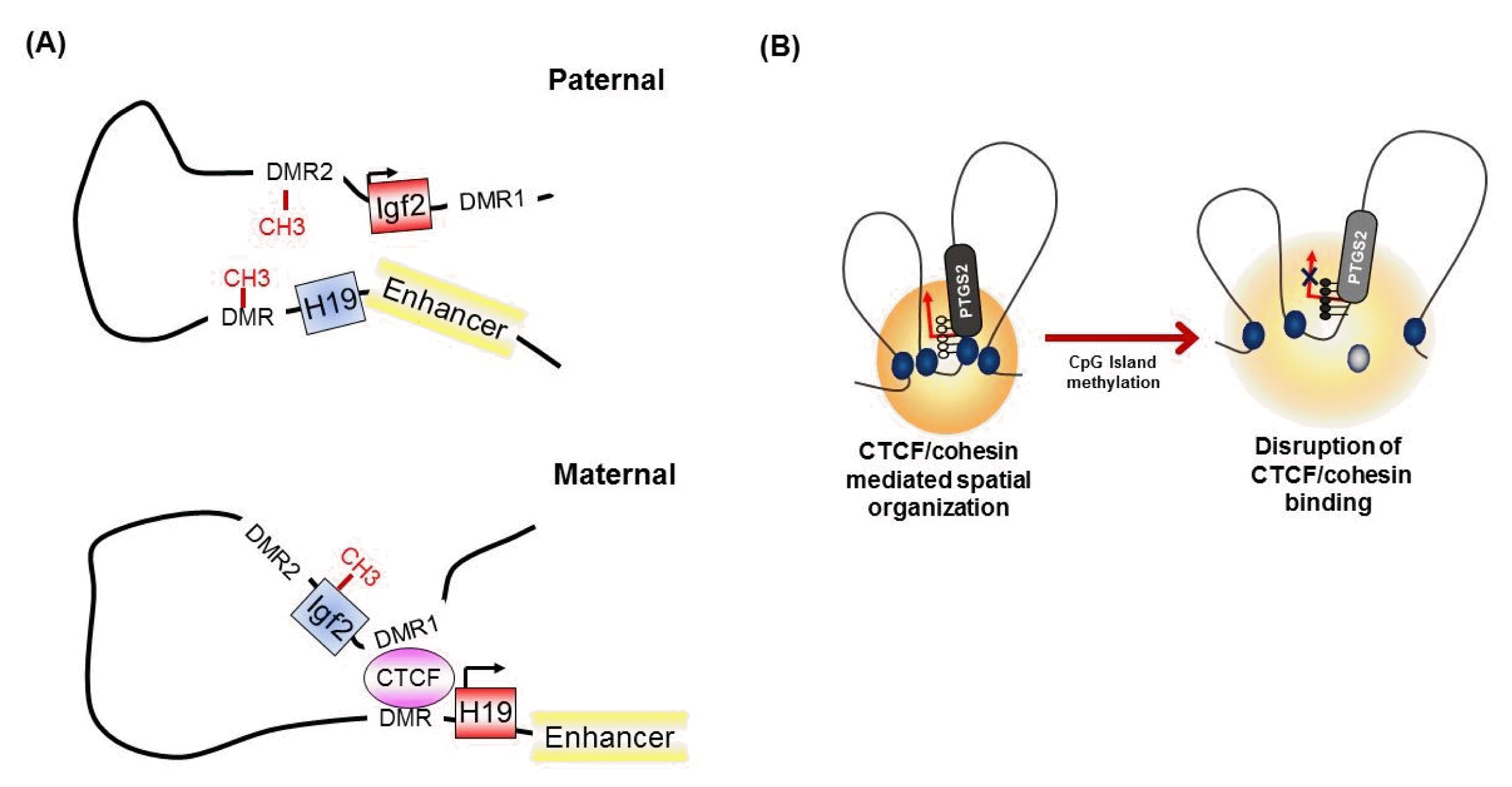
(A, B) Methylation-sensitive binding of CCCTC-binding factor (CTCF).
Abnormal DNA methylation patterns of CTCF-binding sites are associated with transcriptional regulation of tumor suppressor or oncogenic genes in several human cancers [44]. CTCF plays an essential role in maintaining INK/ARF gene expression and disruption of its binding by DNA methylation contributes to the epigenetic silencing of INK/ARF genes in human breast cancer cells [45, 46]. Epigenetic inactivation of RASSF1A and CDH1 also correlates with the epigenetic alteration of CTCF-recognition sites in human breast cancer [46]. Conversely, in one study, aberrant DNA methylation led to the prevention of CTCF-mediated silencing of the BCL6 gene, thus increasing oncogenic BCL6 expression in lymphoma [47].
The concept that the methylation-sensitive binding of CTCF controls gene expression by changing the chromatin architecture has been supported by the finding that CTCF alters the chromatin architecture [8]. For instance, in the Igf2/H19 locus, Igf2 imprinting on the maternal allele is performed by perturbing the proper long-range chromatin interactions between the Igf2 gene and a distal enhancer through the formation of chromatin loops mediated by CTCF (Fig. 3A) [38, 39]. However, on the paternal chromosome, CTCF enrichment at the DMR and insulator looping are prevented by DNA methylation, thus ensuring physical interaction between the Igf2 gene and the distal enhancer and inducing the exclusive expression of the paternal allele. Similarly, nucleotide excision repair factor-mediated DNA demethylation at the promoter region induces the enrichment of CTCF and consequently the formation of a looping structure and controls gene expression at the RARβ2 locus [48]. We also found that epigenetic silencing of PTGS2 correlates with the loss of CTCF binding by DNA methylation at the promoter region, thereby producing an inappropriate higher-order chromatin structure in human gastric cancer cells (Fig. 3B) [49].
Somatic mutations at CTCF-binding sites
In several studies, somatic mutations at the coding region of the CTCF gene were detected in acute leukemia and individuals with intellectual disability [50–52]. However, a high frequency of recurrent mutations in the CTCF-binding site has been more profoundly found in human cancer [53]. Unsurprisingly, single-nucleotide polymorphisms also confer disease susceptibility in humans by decreasing the methylation level at differentially methylated CTCF-binding sites such as rs2334499 in the 11p15 region [54]. Since genetic and/or epigenetic alterations frequently occur in the CTCF anchor region in various human cancers [7, 8, 55], these mutations can influence gene expression and tumor progression by abrogating the CTCF-mediated spatial folding of chromosomes [56].
Architectural role of CTCF
There is direct evidence that CTCF can physically interact with other transcriptional regulators, such as the zinc finger protein Yin Yang 1 (YY1), as an X chromosome binary switch [57]. CTCF also forms a complex with the SNF2-like chromodomain helicase protein (CHD8) [58] and the methyl-CpG–binding protein Kaiso [59] through the zinc-finger domain [10]. Thus, CHD8 enhances insulator activity, whereas Kaiso has a negative effect on the CTCF-mediated enhancer blocking activity [58, 59].
Interestingly, the C-terminus of CTCF preferentially interacts with the STAG1 or STAG2 subunit of cohesin [60]. Furthermore, recent genome-wide studies mapping the binding sites of CTCF revealed that CTCF often colocalizes with the cohesin complex throughout the genome [61–63]. Thus, CTCF is generally thought to be required for the localization of cohesin at its binding sites [64].
The cohesin complex
Cohesin as a sister chromatid cohesion molecule
Cohesin, a large ring-shaped molecule that can bind DNA strands, is a multi-subunit protein complex composed of two structural maintenance of chromosomes (SMC) molecules, SMC1 and SMC3, either stromal antigen (STAG) STAG1 or STAG2, and the kleisin subunit RAD21 (Fig. 4) [9]. Cohesin is required to mediate sister chromatid cohesion for proper chromosome segregation in the S phase until cell division [65]. The cohesin complex was also found to be involved in efficient DNA DSB repair [64]. Since cohesin is important for holding sister chromatids together following DNA replication, mutational inactivation of the cohesin complex causes genomic instability and aneuploidy during cell cycle progression in human diseases [11]. For example, Cornelia de Lange syndrome, a rare autosomal-dominant developmental disorder, is caused by mutation of SMC1, SMC3, Rad21, NIPBL, or HDAC8, which encode core components of the cohesin complex or proteins that interact with this complex [66, 67]. Somatic mutations in the cohesin subunits have also been frequently found in several different human tumor types [68–71].
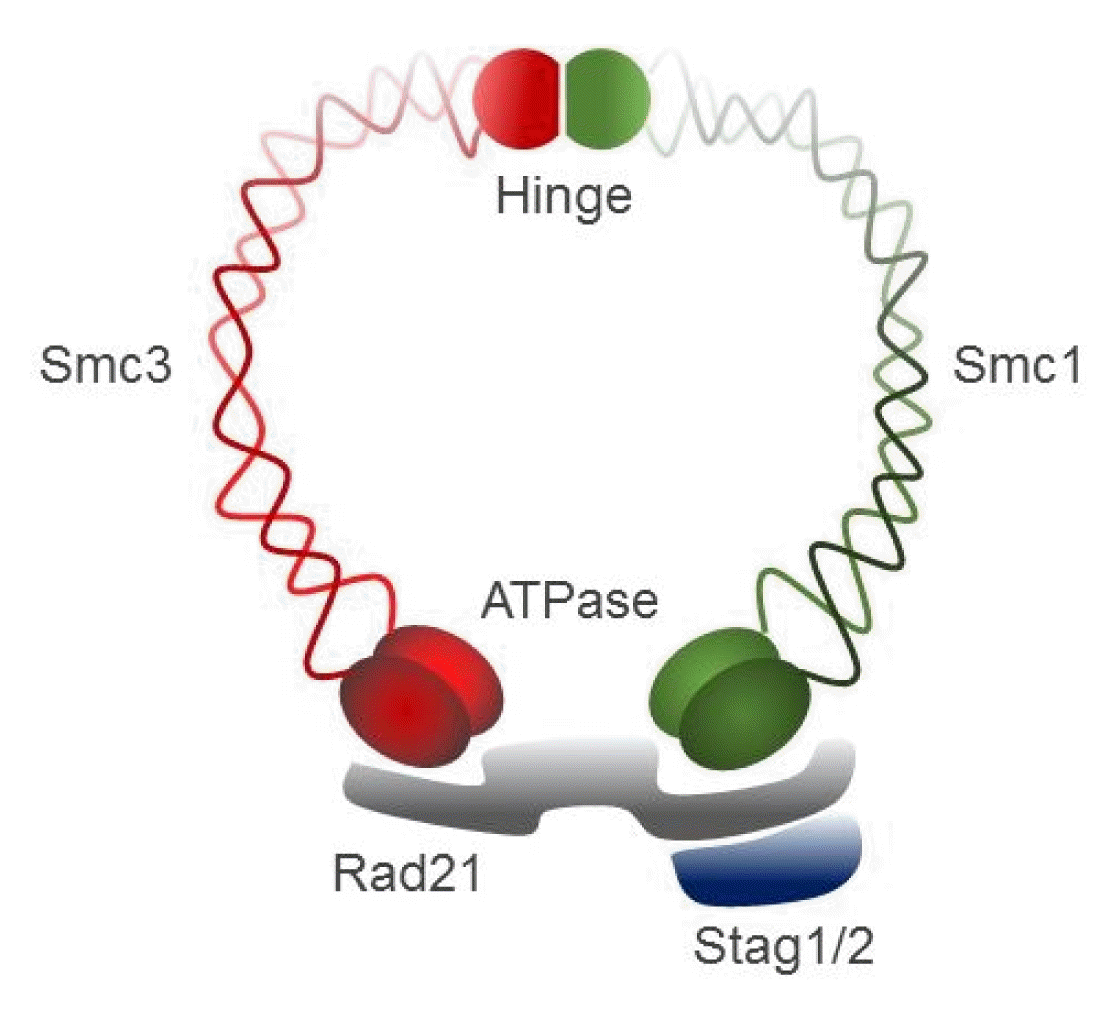
A schematic representation of the cohesin complex. Smc, structural maintenance of chromosomes; Stag, stromal antigen.
Cohesin in transcriptional regulation
Although its role in chromatid cohesion during mitosis is well established [65], cohesin was also found to bind thousands of sites on interphase chromosomes [4]. Indeed, cohesin interacts with the Mediator complex, a transcriptional coactivator [72], and co-occupies enhancer and promoter regions with it to regulate tissue-specific gene expression [4]. High-throughput chromatin immunoprecipitation–sequence analyses also revealed that cohesin remains bound at the transcription factor-binding sites through replication to facilitate the re-establishment of transcription factor clusters after DNA replication and cell division [12]. This suggests that the cohesin complex acts as a transcriptional regulator in cellular proliferation, differentiation, and development [8].
Colocalization of cohesin with CTCF
CTCF was originally known as a cohesin loading factor [10, 64] because genome-wide studies revealed that cohesin globally colocalizes extensively with CTCF throughout the genome [61, 62, 73]. However, CTCF depletion did not completely impair the entire association of cohesin with chromatin [61, 73–75]. Instead, depletion of CTCF was shown to reduce the enrichment of cohesin at only a small proportion of cohesin-binding sites [61, 74, 75], indicating that CTCF facilitates the distribution of cohesin to specific sites on chromosomes [8]. In contrast, the depletion of RAD21, a core subunit of the cohesin complex [9], does not disrupt the enrichment of CTCF, suggesting that CTCF binding is independent of the presence of cohesin on chromatin [8]. In this context, the following question arises: What is the essential role of cohesin at CTCF-binding sites?
Role of CTCF/cohesin in genome folding
Apart from its major function in sister chromatid cohesion, it has recently been shown that cohesin acts in concert with CTCF to affect higher-order chromosome architecture by forming long-range chromosomal interactions in many developmentally regulated gene families [76]. For example, cohesin has been shown to play a critical role in maintaining CTCF-mediated higher-order chromatin conformation at the β-globin and Igf2/H19 loci [37, 39, 41, 74]. CTCF and cohesin also stabilize the rearrangement of Igh and T-cell receptor loci via long-range chromatin interactions [41, 77, 78].
Although it is not yet clear how CTCF/cohesin mediate chromatin looping, CTCF may first bind between two CTCF sites and form a complex with cohesin through its C-terminal region [60, 79, 80]. Considering the evidence that cohesin can tether DNA molecules together [81], a study appeared to show that cohesin stabilized long-range chromatin interactions by anchoring DNA strands together within a closed ring structure among CTCF/cohesin localization sites [11, 64, 76]. Interestingly, CTCF/cohesin-mediated chromatin looping preferentially occurs between CTCF sites with convergent CTCF DNA motifs [82]. Thus, inverting one site of convergent CTCF-binding sites changes the chromatin architecture and can alter gene expression [5, 83].
CTCF/cohesin-mediated abnormal higher-order chromosome structure during tumor development
Dysregulation of the components of the cohesin complex might promote genomic instability by perturbing proper long-range chromatin interactions, which can confer the spatial proximity required for the rejoining of DSBs during chromosomal rearrangement [18]. More recent studies support a similar role for the CTCF/cohesin-mediated chromatin loop as a regulator of genome integrity [84]. They found that chromosome loop anchors bound by CTCF and cohesin are vulnerable to continuous DNA breaks and translocation breakpoint regions in various cancers are enriched at these loop anchors [84]. Similarly, we found that cohesin-mediated chromatin organization and DNA replication are important for stabilizing gene amplification in cancer cells with chromosomal instability (Fig. 5) [85]. Although a high frequency of recurrent mutations and deletions of the components of the cohesin complex was identified in human diseases [70, 71, 86], aberrant overexpression of the cohesin complex [9] was also frequently detected in various human malignancies [85]. Interestingly, we found that overexpression of the cohesin complex in mesenchymal cancer cells induces mesenchymal to epithelial transition–specific expression patterns and dynamic cohesin-mediated chromatin structures are responsible for the initiation and regulation of essential epithelial to mesenchymal transition–related cell fate changes in human cancer (Fig. 6) [87].
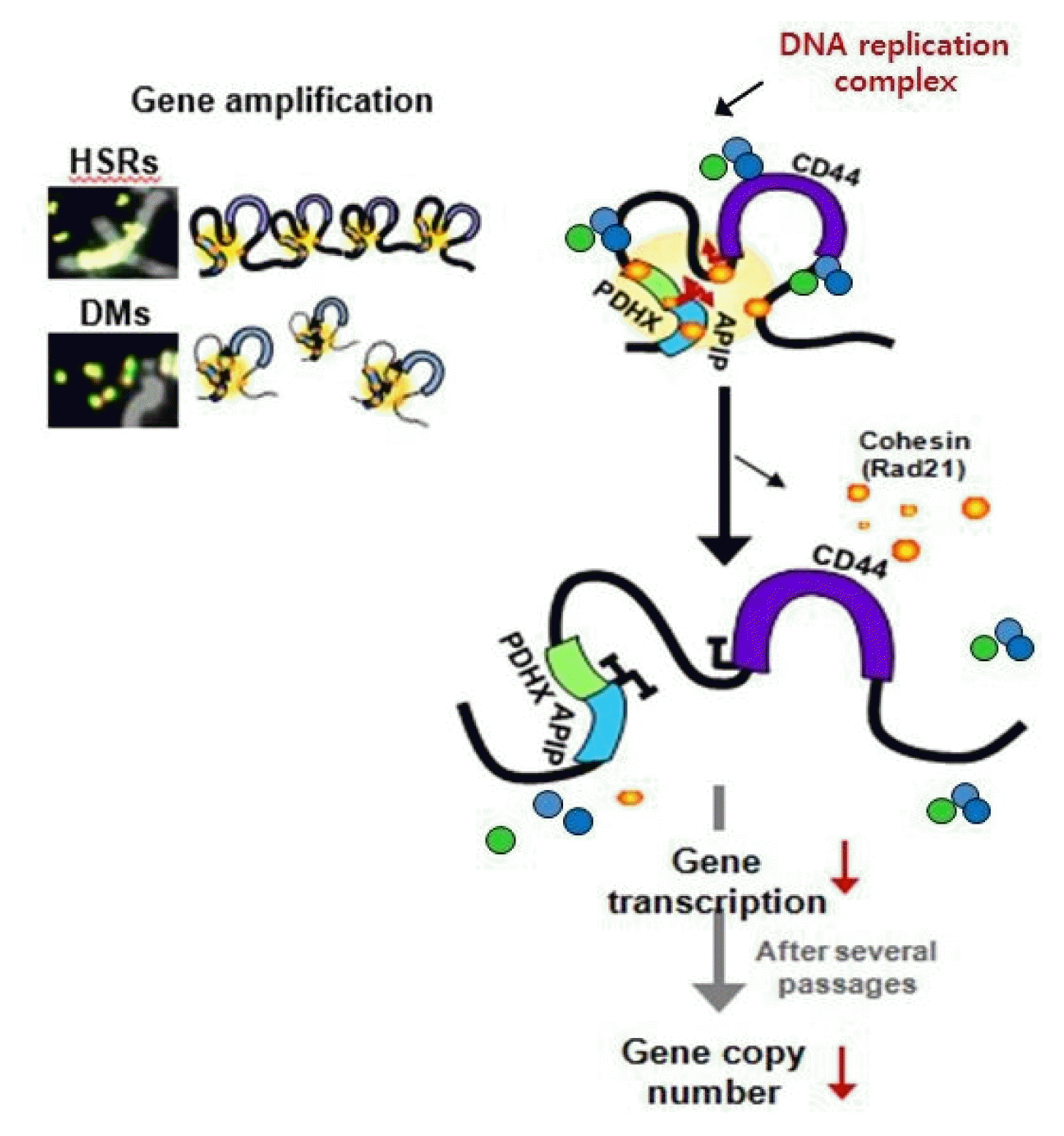
Cohesin-mediated higher-order chromatin structures are important for the expression and presence of high-level gene amplification in cancer cells with chromosomal instability. HSRs, homogeneously staining regions; DMs, extrachromosomal double minutes.

Cohesin-mediated dynamic chromatin architecture of the TGFB1 and ITGA5 genes associated with epithelial to mesenchymal transition (EMT) plasticity. MET, mesenchymal to epithelial transition.
An increasing amount of recent evidence has indicated that CTCF and cohesin are enriched at TADs [6, 15]. Accordingly, cohesin-associated CTCF loops occur within TADs and enhancers generally interact with genes within these loops [8]. Interestingly, recurrent mutations occur frequently within CTCF anchor sites adjacent to oncogenes or cancer-associated genes [55]. Mutation in the isocitrate dehydrogenase (IDH) gene promotes susceptibility of the CTCF-binding sites to DNA methylation and the loss of CTCF binding resulting in the disruption of TAD organization in human gliomas [88]. Somatic mutations change oncogene-containing insulated neighborhoods, thereby allowing improper activation of proto-oncogenes by enhancers located within different TADs [89]. Furthermore, disruption of CTCF-mediated TAD formation by human noncoding disease variants elicits pathogenic phenotypes, providing the mechanistic linkage between spatial genomic organization and genetic alterations that influence gene expression [90].
Conclusion
Over the past few years, substantial progress has been made in understanding three-dimensional genome architecture. However, its role in cancer remains incompletely understood. An intriguing issue in this context is that, although most topological boundaries are enriched for the binding of CTCF, only 15% of CTCF-binding sites are located within TAD boundaries [15], suggesting that additional factors other than the CTCF/cohesin complex might be required to establish the topological domain structure of the genome [17]. Further work is needed to clarify the mechanisms underlying this level of chromosomal organization, and to what extent it generally contributes to the transcriptional regulation of genes during tumorigenesis.
While increasing evidence has recently indicated that enhancers are located near oncogenic genes and exhibit a large number of variants associated with diseases [91], a more complete understanding of how epigenetic alteration of enhancers directly participates in the development and onset of genome reorganization during tumor progression remains to be obtained. Based on this point, recently developed CRISPR/Cas9-based epigenome-editing technology has attracted considerable interest because this approach can acutely modify the epigenetic landscape of specific regulatory elements [92]. For example, targeted editing of the DNA methylation status of CTCF-binding sites changes CTCT recruitment, thereby altering the expression of genes by influencing the organization of higher-order chromatin structures [93]. Furthermore, a high-throughput CRISPR activation system was also used to reveal how noncoding variation associated with human immune dysfunction alters stimulation-dependent enhancer function [94]. Thus, highly specific CRISPR/Cas9-based epigenome-editing technology may serve as an attractive treatment option for epigenetic-based cancer therapies in the coming years.
Acknowledgments
This research was supported by a grant of the Bio & Medical Technology Development Program of the National Research Foundation funded by the Ministry of Science and ICT, Republic of Korea (NRF-2016M3A9B6026918).
Notes
Authors’ contribution
Conceptualization: SHS, TYK
Data curation: SHS, TYK
Formal analysis: SHS, TYK
Funding acquisition: TYK
Methodology: SHS, TYK
Writing – original draft: SHS, TYK
Writing – review & editing: SHS, TYK