Introduction
Osteoporotic fractures (OFs) result from a decrease in bone strength, which can be induced by low bone mass and microarchitectural deterioration in bone tissue [1]. OFs are the critical hard outcome of osteoporosis, a disease that affects more than 75 million people in the United States, Europe, and Japan. With a lifetime fracture risk of 30% to 40% (vertebral or non-vertebral fractures), osteoporosis has an incidence rate similar to that of coronary artery disease. Furthermore, with the exception of forearm fractures, OFs are associated with increased mortality. Most fractures cause acute pain and lead to patient hospitalization, immobilization, and slow recovery [2,3,4].
Genetic studies have suggested a correlation of genetic variations with OF and its related traits. First, a candidate gene study has reported that common non-synonymous variants in low-density lipoprotein receptor-related protein 5 (LRP5) are consistently associated with bone mineral density (BMD) and fracture risk [5]. Furthermore, meta-analyses of genome-wide association studies (GWASs) have identified single-nucleotide polymorphisms (SNPs) located in more than 56 loci independently associated with BMD, and some of these studies have also found associations with fracture risk [6,7,8,9,10,11,12,13].
Copy number variations (CNVs) also have shown associations with OF. A genome-wide CNV study performed in a Chinese population identified a deletion variant of UDP glucuronosyltransferase 2 family, polypeptide B17 (UGT2B17) in chromosome 4q13.2 associated with OF [14]; however, this variant was not replicated in a study of Caucasian women [15], possibly showing ethnic specificity. Additionally, a rare deletion variant located on chromosome 6p25.1 showed an association with the risk of OF in a Dutch population [16].
Particularly, chromosome 20q13.12 is known as an enriched region of histone modifications. Many genes located on chromosome 20q13.12 have enriched levels of histone modifications, such as di-methylated lysine 4 of histone 3 (H3K4me2) and tri-methylated lysine 4 of histone 3 (H3K4me3), in their promoter regions [17]. H3K4me2 and H3K4me3 regulate the expression of the Runx2 gene, which encodes a transcription factor controlling bone development and osteoblast differentiation [18]. Although there is no previous study on an association between chromosome 20q13.12 and bone-related traits, an SNP in another 20q region (rs4811196 in CTNBL1, located on 20q11.23) has been reported to be associated with BMD [19].
In this study, we conducted an association study in the Korean population to identify new susceptibility loci for the risk of OF. We identified 8 CNV regions associated with the risk of OF through a statistical analysis. Among the 8 regions, we selected and validated the existence of one CNV by quantitative PCR.
Methods
Study subjects and diagnostic criteria for OF
A total of 10,038 participants from the Korea Association Resource (KARE) were recruited. Among the recruited individuals, 1,537 subjects, consisting of 299 OF cases and 1,238 normal controls, who agreed to an X-ray examination were genotyped with the NimbleGen HD2 3 ├Ś 720K comparative genomic hybridization array (aCGH). This study was approved by the Institutional Review Board of the Korea Centers for Disease Control and Prevention, and written informed consent was provided to all participants, including cases and controls. The clinical characteristics of the study participants are summarized in Table 1. We used low trauma fracture events to distinguish between fracture and non-fracture groups of the KARE study. Fractures were only included if they had occurred with low trauma (e.g., fall from standing height or less) at any of six sites (hip, wrist, humerus, rib, pelvis, and vertebra) after the age of 40 years. Fractures clearly caused by high trauma events (e.g., motor vehicle accidents, violence, or falls from more than the standing height of the individual) were excluded.
Platform of CNV genotyping
We used the Roche NimbleGen 3 ├Ś 720 K aCGH platform for CNV calling. This platform was designed as a whole-genome and CNV-focused type, composed of a total of 720,000 probes. The median inter-probe spacing of the backbone was <5 kb. DNA extracted from the NA10851 cell line was utilized as a reference for the aCGH to yield the signal intensity ratio with hg18/NCBI build 36. All of the samples in our study satisfied experimental control metrics, such as chromosome X shift and mad.1dr, with NimbleScan v.2.5 to adapt systemic biases in the microarray experiment. The signal intensity ratio of each probe was converted into log2 scale with the positions of the probes after a quality control process.
CNV discovery
CNV discovery consisted of two steps: CNV calling and determination of the CNV region. For CNV calling, we used a package, called ŌĆ£Genome Alteration Detection Analysis (GADA),ŌĆØ which implements a flexible and efficient pipeline to detect copy number alterations from microarray data [20]. To avoid a limitation caused in the single detection tool, we found the best parameter (T = 10, alpha = 0.2, and Min-SegLen = 10) that compared a known CNV region and CNVs tested using several parameters [21]. CNV was employed as the average log2 ratio of ┬▒0.25 of probes in all individual. To estimate CNV genotypes, a CNV region was defined with a log2 ratio identified between the reference sample and test sample in the calling process. It is hard to define exact genotype cluster of a CNV detected in a single individual. To discover CNV regions, we employed CNVs that were called in over three individuals [22].
Genotype estimation
CNV genotypes consist of zero copies, one copy, and two copies. To do this, we used an R package, named ŌĆśCNVtools,ŌĆÖ with default parameters to estimate CNV genotypes [23]. CNVtools is a mixture model for one-dimensional CNV data summary that can separate samples into each CNV genotype. To estimate genotype, we employed the linear discrimination function, which uses a raw signal and pca signal calculated from the average log2 ratio of all individuals using CNVtools. CNV genotypes consist of three genotype groups: ŌĆ£single-class,ŌĆØ ŌĆ£multiple-class,ŌĆØ and ŌĆ£ambiguousŌĆØ (Supplementary Fig. 1) [24,25]. We performed an association study with only well-clustered ŌĆ£multiple-classŌĆØ CNV regions.
Statistical analysis
Logistic regression analysis was performed using R package version 3.0.2 to find significant association signals between CNV loci and OF risk. Age, height, and weight were adjusted as covariates to calibrate the statistical significance. Genotypes of each CNV were coded as 0 (homozygous deletion) and 1 (heterozygous deletion) for two-class CNV loci. In the case of three-class CNVs, their genotypes were coded as 0 (homozygous deletion), 1 (heterozygous deletion), and 2 (normal).
Validation of CNVs
We carried out quantitative PCR using the TaqMan Copy Number Assay (Life Technologies, Foster City, CA, USA) according to the manufacturer's protocols to verify estimated CNV regions. In total, seven pre-designed and one custom designed probe was used to validate the existence of the CNV. Validation samples, including cases and controls, were randomly selected from each estimated genotype cluster. All validation experiments were replicated 3 times to increase the accuracy of the validation. The copy number in each individual was calculated with Copy Caller v2.0 using the comparative threshold cycle (CT) method according to the manufacturer's protocols.
Results
Characteristics of study subjects
A total of 1,537 Korean individuals, consisting of 299 OF patients and 1,238 normal controls, participated in this study (Table 1). There were 415 males and 1,122 females in the overall population. The ratio of females in controls was slightly higher than in the cases (70% in cases vs. 72% in controls, data not shown). Mean age, height, weight, and body mass index showed no significant differences between cases and controls.
Selection of candidate CNV regions
To select candidate CNV regions, we manually performed a visual inspection to classify well-clustered ŌĆ£multiple-classŌĆØ regions into genotype groups. In total, 3,660 multiple-class CNV regions were selected for the association study. Of the 3,660 CNV regions, 518 CNV regions were two-class regions (one and two copies) and 3,142 were three-class regions (zero, one, and two copies) (Supplementary Fig. 1).
Association analysis of CNV regions with OF risk
We performed logistic regression analysis to evaluate associations between identified CNV regions and OF risk. As a result, we identified a total of eight CNV loci associated with OF (p < 0.05) (Table 2)ŌĆödistributed on chromosome 4q13.1, 4q13.2, 9q34.2, 13q12.2, 14q24.3, 14q31.3, 20p13, and 20q13.12. The OF-associated loci were located in intergenic regions near SRIP1; TMPRSS11E; POLR1D; LOC-100421611; a family of long non-coding RNAs (lncRNA), LINC01260; and genic regions of ABO, ELMSAN1, and SIRPA. The base-pair lengths of the eight regions ranged from 1.9 kb to 18.6 kb. The CNVs in ABO and SIRPA included both exonic and intronic regions of each gene, whereas the CNV locus in ELMSAN1 included only intronic regions.
Validation of a CNV region on chr20: 42739446ŌĆō42741539
We selected one CNV region located on chromosome 20q13.12ŌĆöchr20: 42739446ŌĆō42741539ŌĆöor validation based on position and expected biological function. The allele frequencies of chr20: 42739446ŌĆō42741539 are indicated in Supplementary Table 1 and Supplementary Fig. 2. First, we conducted in silico verification for the existence of the CNV region using web-based public data. Many previously reported CNVs, including the chr20: 42739446ŌĆō42741539 region, were identified using the UCSC Genome Browser (http://genome.uscs.edu/cgi-bin/hgGateway) (Supplementary Fig. 3). Also, frequencies of reported CNVs evaluated in various ethnic groups were presented in the Database of Genomic Variants (DGV, http://dgv.tcag.ca/dgv/app/home) (Supplementary Table 1).
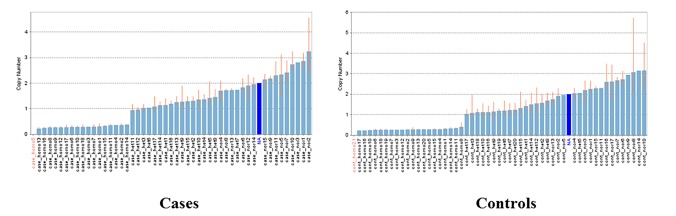
To experimentally verify the accuracy of the CNV, we performed quantitative PCR. The position of the CNV probe was defined based on hg19/NCBI build 37. The probe ID used for the TaqMan genotyping assay of the validated CNVs is listed in Table 3. Applications of a positive predictive value as a measurement standard of accuracy indicated the proportion of positive results in the validation of the CNV region. The positive predictive value of the verified CNV region evaluated in our validation was 0.82. Fig. 1 shows the genotype of the identified CNV region, verified by quantitative PCR.
Discussion
In this study, we performed a GWAS to investigate the associations between identified CNV regions and the risk of OF. A total of eight CNV loci were identified to be nominally associated with OF. For validation, we selected one CNV region based on position and expected biological meaning among the eight loci. The CNV, which is located on chromosome 20q13.12 (intergenic region near LINC01260) was experimentally validated through quantitative PCR.
An lncRNA is defined as a non-coding transcript having a length of more than 200 nucleotides [26] and is considered ŌĆ£junkŌĆØ that is non-functional initially [27]. However, the functional roles of lncRNAs have now been identified, such as key regulators of transcriptional and translational products that affect cell functions [28,29,30,31]. lncRNAs play a role in epigenetic effects, such as histone modification (e.g., methylation), which regulates gene expression [32]. A significant histone modification, monomethylation of lysine 4 on histone H3 (H3K4me1) in human mammary epithelial cells, was predicted in our identified CNV region near an lncRNA, LINC01260, using University of California Santa Cruz (UCSC) Genome Browser (http://genome.uscs.edu/cgi-bin/hgGateway) (Supplementary Fig. 4). A previous genome-wide study has identified several runs of homozygosity (ROHs) associated with BMD and observed the enrichment of H3K4me1 in two ROHs, ROH15q22.3 and ROH1p31.1, by an analysis of histone modification marks in the Gm12878 cell line [33]. Taken together, our identified CNV region on 20q13.12 might have a putative effect on the risk of OF by histone modification of the promoter region of LINC01260.
In addition, a CNV on chromosome 20q13.12 is located in a region about 35 kb apart from the WISP2 gene. The WISP2 gene has some evidence of functions associated with bone-related traits. WISP2, also known as CCN family member 5 (CCN5) or connective tissue growth factor like (CTGF-L), was identified as a member of the CCN family, downstream of the WNTŌĆō╬▓-catenin signaling pathway, using subtractive hybridization between WNT1-transformed mammary epithelial cells and parental cells [34]. The WNTŌĆō╬▓-catenin signaling pathway has been suggested to be a major component in the accrual, regulation, and maintenance of bone mass [35]. WISP2 consists of 3 domains: an insulin-like growth factor binding domain, a von Willebrand Factor type C motif, and a thrombospondin type I repeat. The mRNA of WISP2 is expressed in primary human osteoblasts, fibroblasts, ovary, testes, and heart. WISP2 protein is secreted from primary human osteoblasts and fibroblasts [36]. A previous study has shown the relatedness of increased mRNA expression of WISP2 with rheumatoid arthritis and osteoarthritis [34]. Furthermore, the WISP2 gene has been reported as a candidate gene that is associated with an OF-related trait, BMD [37].
WISP2 expression is affected by the action of LRP5. LRP5 has been known to play a role in signaling through WNTŌĆō╬▓-catenin and acts as a co-receptor with the frizzled family of receptors [38]. Previous research has reported that the LRP5 mutation (G171V), inducing the enhancement of structural strength and material properties of skeleton and bone mass, is correlated with increased expression of WISP2 [38]. Also, WISP2 is suggested to be involved in the frizzled receptors/LRP5/6 pathway by phosphorylation of LRP5 [39]. Considering the previously reported functions and interactions with LRP5 in the WNTŌĆō╬▓-catenin signaling pathway, WISP2 could be an important component for bone formation or strength, which might affect the risk of OF.
In conclusion, we identified a total of 8 CNV regions associated with OF in the Korean population. Among the 8 CNVs, one locus, located on chromosome 20q13.12, was selected for validation and verified by quantitative PCR. Although further study in a larger number of study subjects is needed, to our knowledge, the current report is the first to investigate the association with the risk of OF. Our findings from this study could provide new insights into the genetic factors associated with OF risk.