Introduction
Artificial selection is an aspect of the process of domestication development [1] that is reflected in the evolution of domestic animals. Positive selection increases the fitness of adaptive traits [2]. Previous research has demonstrated that the selection and mating of domestic animals is determined by human-generated pressures, and the genetic differences resulting from artificial selection have led to economic development [3]. Among domestic animals, the pig has experienced a particularly long history of haplotype changes through artificial selection in the process of domestication [2,4]. For most of their history, studies have found positive selection in Yorkshire, Landrace, and Duroc to be associated with specific genes related to lactation [5,6], reproduction [7], meat quality [8], and growth [3] trait. Given the importance of these traits to the pig farming industry, it is necessary to detect the signals of positive selection in pigs.
Identifying genomic selection using high-density single nucleotide polymorphism (SNP) arrays or sequencing data is useful for the discovery of putative trait-related genes. A variant under selection pressure between and/or within populations will show an increase of the frequency of particular alleles or linkage disequilibrium (LD) of SNPs. Selective sweep can cause genetic differentiation across populations [9]. The cross-population extended haplotype homozygosity (XPEHH) [10] and cross-population composite likelihood ratio (XPCLR) [9] tests are widely used to search for signals of selection between the populations. The XPEHH test detects the occurrence of selection based on measuring LD, while the XPCLR test considers the spatial patterns of allele frequencies of SNPs [9]. The XPCLR test is a likelihood method that uses differentiation of multi-locus allele frequencies between two populations to detect selective sweep. Selection with significant signals can break up the haplotype structures more rapidly than mutation or recombination processes [11].
In this study, the XPEHH and XPCLR between-population methods were implemented to detect positive selection in three pig breeds through resequencing using the Illumina HiSeq2000 platform (Illumina, Inc., San Diego, CA, USA). To further explain the biological implications of positively selected genes, functional enrichment analysis was performed.
Methods
Sampling and whole-genome sequencing
We used genomic DNA samples gathered from 12 males and 12 females of three pig breeds: Yorkshire, 2 males and 3 females; Landrace, 7 males and 6 females; and Duroc, 3 males and 3 females. Blood samples were collected for DNA extraction. Sample collections and DNA quality check procedures were performed according to the manufacturer's instructions. Next, we constructed genomic DNA libraries for each sample using TruSeq DNA Library kits (Illumina). The paired-end library was sequenced on an Illumina HiSeq 2000 sequencing platform.
The pair-end sequence reads were aligned to the reference pig genome sequence from University of California, Santa Cruz (UCSC; http://genome.ucsc.edu/; susScr3) using Bowtie2 with the default setting. We used the following open-source software: Bowtie2, Picard tools 1.94 (http://picard.sourceforge.net), Samtools 0.1.19 [12], Genome Analysis Toolkit (GATK) 2.6.4 [13], and VCFtools 4.0 [14] for resequencing data processing and SNP calling. Substitution calling was performed using GATK Unified-Genotyper. We phased the haplotypes for the entire pig populations using BEAGLE [15]. Picard tools was used for duplicate read removal and all mate-pair information confirmation. Samtools was used for indexing the results from bam files and calculating the mapped reads using the flagstat option. GATK was used for realignment and SNP calling from resequencing data, and VCFtools was used when VCF files were handled. After filtering, non-biallelic SNPs were excluded.
Detection of selective sweep
To detect selective sweep, we implemented two between-populations methods (XPEHH and XPCLR) between each pairwise breed contrast. These statistics used the phased data based on the SNP genotypes obtained from the wholegenome resequencing. For each contrast, XPEHH and XPCLR were implemented across all SNPs between the three pig breeds to identify potential positive selection. We used a p-value of <0.01 for XPEHH and the top 1,000 genes in XPCLR as the cutoff criteria. These tests revealed that some SNPs might have been under positive selection in the Yorkshire, Landrace, and Duroc breeds, respectively.
Bioinformatic analysis of genes under positive selection
Based on the findings of positive selection, enrichment analysis was performed to examine the biological functions of genes in detected regions. In this study, regions with SNPs under positive selection were extended approximately 10 kb upstream and downstream [16]. We assembled genes located within the extended region using the RefGene from the UCSC Genome Browser (http://genome.ucsc.edu/; ver. hg19). Gene enrichment analysis was performed GO terms [17], including biological process, molecular function, cellular component, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis [18] using the DAVID (http://david.abcc.ncifcrf.gov/) tool [19].
Analysis of SNP effects
To identify non-synonymous coding SNPs (nsSNPs), we analyzed SNP effects using the SNPeffect database, which detects phenotypic effects of variation (http://snpeffect.vib.be/) [20]. For more exact results, a Fisher's exact test was performed to identify breed-specific amino acids in genes with nsSNPs. The statistical test was used to analyze 2 ├Ś 2 contingency tables composed with two factors. Considering our purpose, one factor was breed information, and the other was amino acid information. From the 2 ├Ś 2 contingency tables, we expected to observe enrichment of specific amino acids in specific breeds at each position. All data parsing and calculations were performed using Python (ver. 2.5) and R (ver. 3.1.2).
Results
Sequencing and alignments
Genome-wide positive selection in pigs was identified using whole-genome resequencing, in which we aligned whole genome reads from three pig breeds, Yorkshire, Landrace, and Duroc, to the pig reference sequence. These breeds have undergone positive selection with strong signals for lactation, reproduction, meat quality, and growth. On average, ~329,482,871 sequences were mapped to the pig reference genome from the University of California, Santa Cruz (UCSC) Genome Browser (assembly ID: susScr3) using Bowtie2. Table 1 provides a summary of the alignments of 24 pig samples. Over 92.8% of all reads were aligned to the pig reference genome. Pig resequencing data with an average read depth of 11.7├Ś were generated using the Illumina HiSeq2000 platform. A total of 18,440,443 SNPs were identified using the GATK.
Selective sweep detected by between-population methods
To detect signals of positive selection in the three pig breeds, two between-population methods (XPEHH and XPCLR) were implemented with phased haplotypes based on pairwise populations of Yorkshire-Landrace (Y-L), Yorkshire-Duroc (Y-D), Landrace-Yorkshire (L-Y), Landrace-Duroc (L-D), Duroc-Yorkshire (D-Y), and Duroc-Landrace (D-L). Here, for Y-L, Yorkshire was regarded as the observed population and Landrace as the reference population. XPEHH and XPCLR scores denote selection that happened in the observed population. As a result, several regions of positive selection were identified at a significance level of p-value < 0.01 [21] in XPEHH, and the top 1,000 genes in XPCLR. Table 2 summarizes the numbers of genes under significant positive selection, which were identified in the six breed pairs (Y-L, Y-D, L-Y, L-D, D-L, and D-Y). For example, for the Y-L breed pair, which detects selection that occurred in the Yorkshire population when compared to the Landrace population, 376 and 1,869 genes were under positive selection based on the XPEHH and XPCLR tests, respectively. For the Y-D breed pair, when using the XPCLR test, 1,498 genes were under positive selection with 3,978 SNPs detected in Yorkshire, with Duroc as the reference population. By the consensus set of genes under positive selection identified with both the XPEHH and XPCLR tests, 26, 7, and 14 genes were identified in Yorkshire, Landrace, and Duroc, respectively (Fig. 1). For example, the 26 Yorkshire genes identified by both the XPCLR and XPEHH tests in both the Y-L and Y-D comparisons, and so effectively detected four times.
The biological functions of genes under positive selection
We performed functional enrichment analysis on the following genes under positive selection using DAVID ver. 6.7: ST6GALNAC2 and EPHX1 in Yorkshire; PARK2 in Landrace; and BMP6, SLA-DQA1, and PRKG1 in Duroc. The results revealed enrichment of Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway terms related to metabolism, diseases, genetic or environmental information processing, and organismal systems (Table 3). The gene BMP6 acts as a hedgehog signaling pathway; the hedgehog protein is an important regulator of slow oxidative fiber clustering and is distributed throughout the tissue in pigs. Previous research has reported that BMP6 plays an important role in meat quality [8]. Among the 26 positively selected genes in Yorkshire, the ST6GALNAC2 gene is expressed during lactation, contributing to immune-related functions [5]. Up-regulation of the EPHX1 (epoxide hydrolase 1) gene occurs during mammary gland involution. Nakamura et al. [6] suggested that EPHX1 is involved in the initial stage of involution. The SEMA5A (semaphoring 5A) gene for Yorkshire reported strong candidate gene which affects the milk production traits for economic traits in cattle. The estrogen receptor (ER) gene for Landrace reported that mice lacking ER ╬▒ show severe reproduction [7]. This implies that the ER gene might be a positively selected lactation trait in pigs. The Gene Ontology (GO) terms included immune system process (SLA-DQA1) and metabolic process (PRKG1) for Duroc (Supplementary Table 2). Williams et al. [3] reported that a low level of chronic immune system resulted in greater growth in pigs. These results suggest that the 26, 7, and 14 population-specific genes with significant signals may result from potential positive selection for lactation in Yorkshire, reproduction in Landrace, and meat-quality and growth traits in Duroc.
SNP effects for population-specific genes
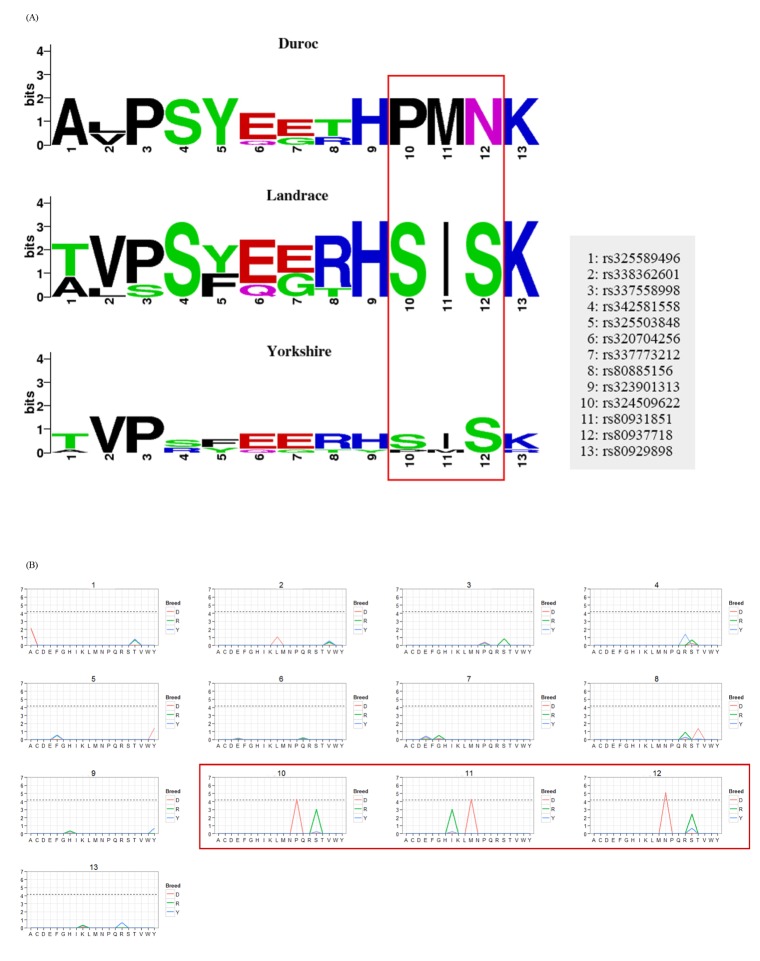
To understand how SNPs related positive selection affect protein function, we analyzed SNP effects using the SNPeffect database. We identified two genes, SLA-DQA1 and EPHX1, with nsSNPs showing evidence of positive selection in D-Y, respectively (Fig. 2, Supplementary Table 1). To obtain accurate results, Fisher's exact test was performed on these two genes. The total numbers of statistical tests performed on the non-synonymous genes were 20 ├Ś 3 ├Ś 13 (number of cases of amino acids ├Ś number of breeds ├Ś number of non-synonymous amino acid positions) for SLA-DQA1, and 20 ├Ś 3 ├Ś 4 for EPHX1. From the enrichment tests, we identified three positions (rs324509622, rs80931851, and rs80937718) that were significant under a Bonferroni-adjusted p-value of <0.05 with an alternative hypothesis: the odds ratio is greater than 1 in the SLA-DQA1 gene. The p-values of the three non-synonymous positions were 5.20e-05, 5.20e-05, and 7.43e-06, respectively. As a note, the above three significant enrichment results were observed only in the Duroc breed. The non-synonymous positions showed strong evidence for positive selection between D-Y based on the XPEHH test. As the result, we validated the Duroc-specific enrichment of amino acids affected by non-synonymous SNPs using statistical tests. This result will contribute to improving the meat quality of pigs.
Discussion
Identifying positive selection assists in the exploration of the genetic mechanisms of phenotypic diversity [2]. Selective sweep causes changes in the allele frequencies of SNPs, and the resulting changes in genetic variation can be useful in detecting selective pressure [9]. Although several studies have reported selective sweeps identified using chips such as the Illumina Porcine 60K SNP BeadChip [22] in pigs, few have identified selective sweeps using whole-genome sequencing. Therefore, detecting positive selection in the pig genome appears worthwhile.
Here we implemented two representative between-population methods, XPEHH and XPCLR, to explore positive selection. XPEHH is designed to detect near fixation or fixed selective sweeps by comparing haplotypes between two populations [10]. XPCLR is more robust for SNP discovery bias than allele frequency-based methods [9]. XPEHH tests whether the SNP site is homozygous in one population and polymorphic in another by comparing the extended haplotype homozygosity score of two populations on a core SNP. A negative XPEHH value indicates that selection happened in the reference population [15]. XPCLR was designed as a multi-locus composite likelihood ratio method to avoid the bias influence in SNP discovery. XPEHH and XPCLR find one core SNP and grid window using multi-locus information, and thus these tests identify more significant selection regions than Fst or Tajima's D [23]. Identifying significant genome-wide selection regions with multiple loci using a single-marker Fst test is difficult. Therefore, for multiple SNP methods, we carried out XPEHH and XPCLR and excluded Fst and Tajima's D. XPEHH assumes an approximately normal distribution. Park et al. [16] determined empirical p-values using the top 1% of the significance, and our results show that these significance cutoffs are more reasonable. In addition, positive selection identified by multiple test methods is more convincing.
Bioinformatic analysis of genes under positive selection in this study revealed interesting biological information related to lactation, reproduction, meat quality, and growth. We identified several GO terms using the DAVID functional annotation tool. Immune system process (GO:0002376) was found to be significant in Duroc. Williams et al. [3] reported that a low level of chronic immune system resulted in greater growth in pigs. This GO term suggests that genes with some growth traits may have been under selection in the domestication process. Growth traits are referred to as important indicators of the immune system. This suggests that SLA-DQA1 might play a role in the pig immune system. In addition, BMP6 and PRKG1 in Duroc had biological pathways related to meat-quality traits. Fonseca et al. [8] reported that the BMP6 gene plays a role in increasing the quality of meat juiciness and tenderness. Lee et al. [22] identified the significant SNPs located in the BMP6 gene related to meat quality through a genome-wide association study using the Porcine 60K BeadChip. Therefore, the SLA-DQA1 and BMP6 genes might be effects of positive selection for growth and meat quality, respectively, in Duroc. In Yorkshire, three genes under significant positive selection were ST6GALNAC2, EPHX1, and SEMA5A; these genes were related to lactation traits in several previous studies. The ST6GALNAC2 gene is expressed during lactation [5], EPHX1 is expressed in the early involution of mammary glands [6], and SEMA5A is reportedly related to milk production traits in cattle [21]. In Landrace, the ER gene was reported to be related to reproduction traits in previous studies. Generally ER refers to either ER ╬▒ or ER ╬▓ and estrogens are involved in differentiation and maintenance of reproductive tissue. Krege et al. [7] reported that mice lacking ER ╬▒ exhibited severe reproductive problems. This implies that the ER gene could be an effect of positive selection for lactation traits in pigs as well as in mice. Our findings show that several significant SNPs were present in genes found to be related to lactation, reproduction, meat quality, and growth traits in previous studies. These results indirectly support the effects of positive selection. Unfortunately, information on many of the genes with significant signals has not yet been analyzed. Based on SNP analyses for positively selected genes, SLA-DQA1 and EPHX1 genes exhibited evidence of positive selection in D-Y, respectively. However, an enrichment test revealed that the three significant amino-acid positions in SLA-DQA1 and EPHX1 were not significant.
Our analyses suggested that several genesŌĆōsuch as EPHX1, PARK2, BMP6, SLA-DQA1, PRKG1, and ST6GALNAC2ŌĆōmight have undergone positive selection. These genes are associated with lactation, reproduction, meat quality, and growth traits. All findings in this study provide useful data for the study of pigs, however our results are preliminary and need to validate these genes in another set of samples.