Introduction
Genome-wide association studies (GWASs) have been a useful tool to identify genetic variants that affect various traits [1]. Numerous novel important genetic variants associated with disease susceptibility have been identified through GWASs [2]. However, genome-wide association (GWA) results for complex diseases can generally explain only a small part of the genetic variants for complex diseases [3]. Generally, a GWA study identifying an association between a trait and a genetic variation may be limited in understanding complex diseases involving multiple functional loci [4].
Recently, many approaches to detect an association between a trait and one or multiple genetic variants have been proposed to study numerous data from GWASs [5]. Inference about linkage disequilibrium (LD), known as non-independent association of alleles at different loci, provides information for the association of genetic variants affecting complex traits [6]. Using LD analysis, it is possible to characterize multiple genomic variants associated with phenotypes in terms of haplotypes. A haplotype defines a combination of phased alleles in a chromosomal region [4]. Thus, haplotype analysis is useful to understand multiloci genetic associations and to identify susceptibility loci for diseases. In spite of the advantages of haplotype analysis, performing an analysis on a genome-wide scale is not simple, due to the uncertainty and complexity of haplotypes [7].
Whole-genome sequencing (WGS) of a reference population may provide insights into the potential causal variants hidden within the LD block of interest. While the 1000 Genomes Project aims to provide such information in worldwide populations or ethnic groups [8], it would be more informative to focus on a reference population on a national scale than on a global scale. Recently, several Korean reference WGS datasets have been published based on next-generation sequencing (NGS) platforms [9]. Here, we tried to construct haplotype blocks affecting body mass index (BMI) in Korean individuals through integrative analysis of NGS and GWA data. We analyzed haplotypes using NGS data only in ranges that were nearby significant single-nucleotide polymorphisms (SNPs) that were identified by GWA results. The analysis may have several advantages in haplotype analysis. The complexity and the time can be reduced by analyzing smaller regions that are known to harbor significant GWA signals. We cataloged all possible 44 haplotype blocks within the LD blocks of significant SNPs for BMI.
Methods
Overall pipeline of the analysis
The NGS raw data were mapped to reference sequences using an alignment tool. The significant SNPs for BMI were selected from the GWA result data. The LD blocks that encompassed the significant SNPs were searched with Haploview [10], and those NGS reads that were mapped to the LD ranges were selected with Samtools. For each LD block, the heterozygous variants from the short reads were phased with Samtools. Finally, the phased haplotypes harboring the significant SNPs were cataloged (Fig. 1).
Genotyping, imputation, and GWA analysis
The Korean samples and genotype data used in this study have been described by Cho et al. [11]. Briefly, through the Korea Association Resource (KARE) project, a total of 10,038 participants were recruited from Ansan and Anseong population-based cohorts, aged 40 to 69. The genotypes that had been measured on Affymetrix Genome-Wide Human SNP array 5.0 (Affymetrix, Santa Clara, CA, USA) were filtered for quality control, resulting in 8,842 samples and 352,228 markers. The genotype data were expanded to a total of 1,827,004 SNPs through imputation with IMPUTE [12] using the Japanese in Tokyo, Japan (JPT)/Han Chinese in Beijing, China (CHB) component of HapMap as the reference.
BMI traits were tested for association by linear regression analysis with an additive model after adjustments for recruitment region, age, and sex as covariates using PLINK. The threshold of genome-wide significance was set at p < 5 ├Ś 10-4. The significant SNPs were clustered into LD blocks (r2 > 0.9) using HaploView [13].
NGS data analysis
We downloaded previously published whole-genome NGS data from the TIARA database, sequenced at Seoul National University College of Medicine (TIARA; http://tiara.gmi.ac.kr) [14]. The short reads from seven samples (AK3, AK4, AK5, AK6, AK7, AK14, and AK20) were mapped to the human reference genome (hg19) using the alignment algorithm in Genomic Short-read Nucleotide Alignment Program (GSNAP) [15]. From the resulting BAM file, we selected the mapped reads within the LD blocks that encompassed the significant SNPs from the BMI GWA result using the Samtools view function [16], creating a separate BAM file for each LD block.
Heterozygous SNP calling and phasing
The heterozygous SNPs were phased using the phase function in Samtools, version 0.1.19-44428cd [17]. The Samtools phase algorithm calls heterozygous SNPs automatically and phases those that segregate on the same DNA fragment as inferred from paired-end read information in the BAM file. We used the default options for the Samtools phase function. We confirmed the heterozygous SNPs through independent calling using the Samtools mpileup function [17]. Whenever an LD block split into multiple phased haplotype blocks, we kept those that encompassed the significant SNPs from the GWAS.
Results
GWA analysis
The genotype data of 8,842 samples in a total of 10,038 participants in the KARE were analyzed to find significant SNPs associated with BMI in Korean individuals. Among the total of 1,827,004 loci, 435 loci were identified using a p-value cutoff (p < 5 ├Ś 10-4) from the GWAS. The default algorithm in the HaploView program for identifying LD blocks was taken from confidence intervals [13]. We set a ┬▒20-kb window option and r2 > 0.9 to capture LD blocks of 435 significant SNPs based on the KARE genotype data. For 435 loci that were significant in the association analysis (p < 5 ├Ś 10-4), 53 LD blocks were identified using HaploView (Table 1). These 53 LD blocks contained all 435 significant loci. Thus, a typical LD block included multiple significant loci.
Phasing short reads
We downloaded whole-genome paired-end NGS data of seven samples from the TIARA database to discover haplotypes with the SNPs associated with BMI within the 53 LD blocks from the GWA analysis. We mapped the short reads to the human reference genome (hg19), and those reads with bad mapping quality (mapping quality <5) were filtered out. The average mapping rate was about 93%. We separated the mapped reads into the 53 LD blocks for each of seven individuals (Table 1). Each group of separated reads was called for variants, and the heterozygous SNPs were phased using the phase function in Samtools. Not all of the SNPs in an LD block were always phased into a contiguous haplotype block. Whenever multiple haplotype blocks were found for a given LD block, we kept only those that encompassed the significant SNPs identified from the GWAS.
Construction of the haplotype blocks
After phasing heterozygous SNPs using Samtools, we searched for phased haplotypes that harbored significant risk alleles for BMI from the GWA analysis results. We found 44 haplotype blocks in 23 of 53 LD blocks. We could not find haplotypes with significant risk alleles in the rest of the LD blocks. We ordered 44 haplotypes and established a catalog for possible haplotype blocks associated with BMI variations in the Korean population (Table 2). For example, AK4 and AK7 share the TCTGAGCC haplotype, which comprises the variants at bps 246142250, 246142279, 246144436, 246146137, 246146178, 246146444, 246148791, and 246149166 in chromosome 1. Bp 57895600 in chromosome 18 has been previously known as rs8089366 (G/T) in the dbSNP database and was a significant SNP for BMI in KARE. On the other hand, bp 57900630, which participated in the same haplotype, has not been registered in the dbSNP database. In Table 2, 16 LD blocks were shared by several individuals, while the rest of the LD blocks were carried by single individuals.
Distribution of haplotype blocks
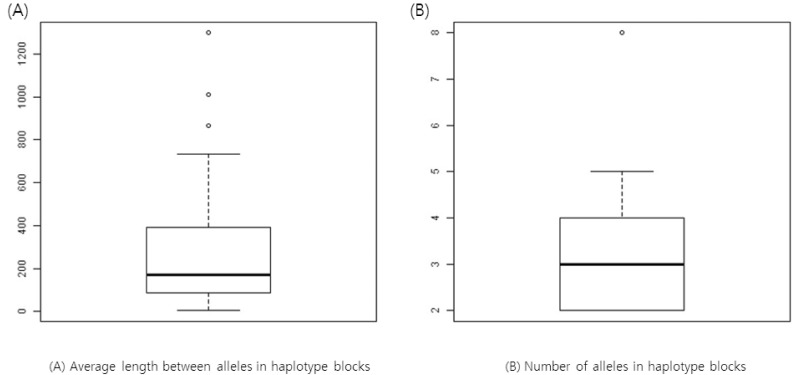
We calculated the average space between alleles in a haplotype block from our results. For example, AK4, AK5, and AK7 share CGCC at bps 182065282, 182065314, 182065341, and 182065351 in chromosome 4 (Table 2). The haplotype block ranges from bp 182065282 through bp 182065351; the length of the haplotype is 69 bp. Since there are four SNPs, the average space is about 17. We plotted the distribution of the average spaces between SNPs within a haplotype (Fig. 2). The number of alleles found for a haplotype is also plotted in Fig. 2.
Discussion
The GWAS is a powerful method to identify genomic variants affecting traits. Many studies have found important SNPs associated with diseases. However, the study is not well suited to identify rare variants or small effects of several SNPs [18]. Here, we tried to find haplotype blocks in 7 Korea individuals that harbor SNPs that are significantly associated with BMI in a Korean population. The significant SNPs for BMI were detected through GWASs, and the haplotypes were discovered by analyzing the NGS data. We selected short reads within the LD ranges that encompassed the significant SNPs from the GWAS results, and the heterozygous variants that were called from those reads were phased, based on the paired-end information. As a result, we detected 44 haplotype blocks harboring SNPs that are significantly associated with BMI in Koreans. The haplotypes may affect BMI in Korea individuals. However, it is not statically powerful, due to the small number of samples (7 individuals) used in this study. Actually, the alleles of a haplotype need to be compared with those of the haplotype of the same range in other individuals to have high accuracy. We could not follow this strategy. For example, most of the haplotypes found by this analysis were discovered in a single individual. In our small dataset, a significant SNP from a GWAS is not likely to be shared by several individuals, because many SNPs have a minor allele frequency of around 10% in a population. This problem may be solved if a large dataset is used. In addition, read depth also reduces accuracy. In Table 2, several haplotypes were identified in a single GWA LD block. These haplotypes could not be joined, as there were not enough short reads that could bridge them using the phase function in Samtools. In addition, AK7 has the GATX haplotype at bps 57903745, 57904011 (rs590654), 57904088 (rs590215), and 57907787 in chromosome 18, and AK4 has XAXC in the same locus. An X represents a missing allele, probably due to low coverage. We were able to recover them, as Samtools was still able to phase them. We filled in these alleles manually to build the haplotype. In Table 2, the green letters represent such alleles that have been filtered by read depth and recovered manually. Although the accuracy is not high, this integrative method has several advantages. First, rare variants can be detected. For example, AK4 has the TT risk allele haplotype at bps 57895600 and 57900630 in chromosome 18. The former was associated with BMI and was registered in the dbSNP database, while the latter was not registered in the dbSNP database. Bp 57900630 is possibly a low-frequency risk allele, which is difficult to be detected by GWAS. Second, we can explain a trait systematically. rs559623 is an SNP (C/A: forward strand) that is associated with BMI. It may act with bp 57844375 in chromosome 18 as CC according to our results (Table 2). Traits can be explained properly by multiple variants. Finally, this method is useful for studying phasing. Many SNPs from an array or NGS reads are unphased genotypes. However, the bulk of SNP information needs to be phased for identifying co-located alleles. While GATK HaplotypeCaller, Samtools Phase, and Beagle are used to phase variants, there are several problems, such as the long execution time and the need for large system memory. In fact, it takes too long to phase entire chromosomes or a single chromosome. However, with this method, it is possible to analyze efficiently with regard to time and memory, as it does not consider reference and genotype data of the entire chromosome. This method focuses only on the data within regions that are nearby important variants from the GWA analysis results. We constructed a crucial haplotype catalog for BMI traits in Korean individuals by integratively analyzing NGS data and GWA analysis data.