Introduction
Cancer cells express programmed death ligand 1 as a signal related to T cell unresponsiveness. Immunotherapies targeting immune checkpoints (e.g., antiŌĆōcytotoxic T lymphocyte associated antigen-4 and antiŌĆōprogrammed death-1 antibodies) are a standard component of care for patients with advanced cancers. Immune checkpoint inhibitors (ICIs) have led to improvements in the survival rate, but only a subset of patients respond to ICIs. Recent studies have reported that the efficacy of ICIs in cancer patients is determined by the T cell inflamed tumor microenvironment [1-3]. The molecular mechanisms of resistance have not yet been elucidated in detail. Nevertheless, previous studies have reported scoring methods for distinguishing nonŌĆōT cell inflamed from T cell inflamed tumors based on gene expression data [4,5].
Unfortunately, general researchers who are unfamiliar with the detailed calculations involved in the equations can experience difficulties using these scoring formulas to conduct analyses. For this reason, we recently developed TcellInflamedDetector, an R package that predicts T cell inflamed tumors when given RNA-sequencing expression data. This package will be beneficial to optimize the selection of patients predicted to benefit from ICIs. TcellInflamedDetector implements the equation developed by Spranger et al. [5] to differentiate nonŌĆōT cell inflamed and T cell inflamed tumor subtypes.
Input Data and Processing
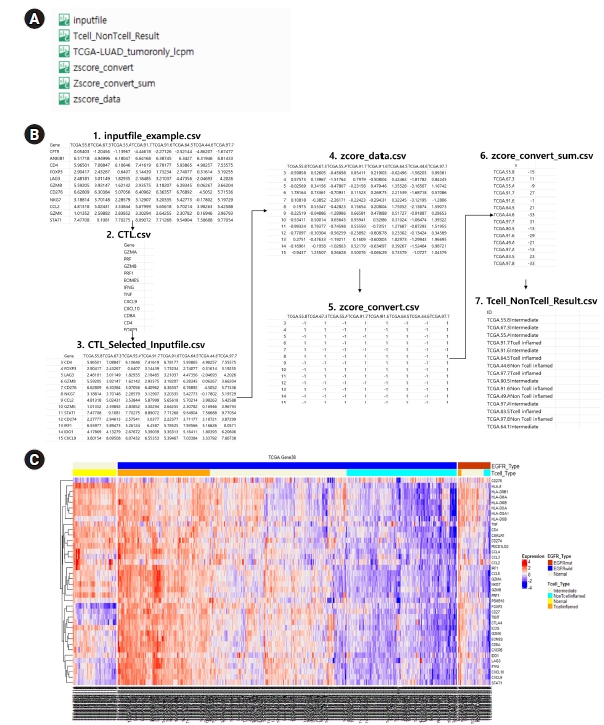
As shown in Fig. 1, TcellInflamedDetector requires RNA-sequencing count input data with genes and sample identifiers. Users follow the steps for data processing that are summarized in the TcellInflamedDetector manual on GitHub [6]. The input CSV file is RNA sequencing log count per million (CPM) data. The count matrix file is converted by EdgeR aveLogCPM() and the calcNormFactor function using the trimmed mean of the m-values method. Users can extract previously established gene signatures indicative of a T cell inflamed tumor microenvironment, which include the cytotoxic T lymphocyte (CTL) signature genes CD8A, CD8B, GZMA, GZMB, and PRF1 using R code [7-10]. The established gene signatures were referenced with the Gajewski T cell-inflamed signature, interferon-gamma related signature, T cell effector signature, and immune cytolytic activity signature [4,5].
Estimating T Cell Inflamed and NonŌĆōT Cell Inflamed Samples
As shown in Fig. 2, gene expression values were converted to a score Si = ┬Ąi ┬▒ ╬▓iŽāi (i = 1, 2, ŌĆ” n), where ┬Ą and Žā represent the mean and standard deviation (SD) of the ith geneŌĆÖs expression across all samples, n is the total number of genes, ╬▓ represents the distance between the ith geneŌĆÖs expression in a sample and the mean in units of the SD (equivalent to a z-score). The threshold for nonŌĆōT cell inflamed and T cell inflamed tumors was ╬▓0 = 0.1. The algorithm is described in detail below:
If the z-score value ╬▓i is greater than the threshold (╬▓0 = 0.1), then +1 is assigned. Otherwise, if the z-score value ╬▓i is less than the threshold (╬▓0 = 0.1), then ŌĆÆ1 is assigned. If the sum of the column of genes with assigned values is greater than half of the number of CTL genes, then the output is a classification of ŌĆ£T cell inflamed.ŌĆØ If the sum of a column of genes with assigned values is less than half of the number of CTL genes, then the classification is ŌĆ£nonŌĆōT cell inflamed.ŌĆØ Otherwise, the sample is classified as ŌĆ£intermediate.ŌĆØ
Users of the R package can obtain results in the format of a .csv file that contains data on the classification of samples as T cell inflamed, nonŌĆōT cell inflamed, and intermediate. If users want to modify the CTL gene list when running the R package, they do not have to modify the complex R code. Instead, they can simply revise the gene list contained in the CTL.csv file.
Output
Five output formats are available: CTL_Selected_Inputfile.csv, Tcell_NonTcell_Result.csv, zscore_convert.csv, Zscore_convert_sum.csv, and zscore_data.csv. Fig. 3 presents examples of the prediction results of T cell inflamed, intermediate, and nonŌĆōT cell inflamed groups. Users can check the expression patterns of specific genes through a heatmap. We also confirmed that T cell inflamed samples showed high expression of T cell effector gene signatures [10].
Finally, we conducted a test to demonstrate our toolŌĆÖs flexibility; we tested it on The Cancer Genome Atlas (TCGA) lung adenocarcinoma RNA-sequencing dataset available through the TCGA Research Network [11]. Each sample was labeled according to the TCGA barcode, which contained gene names. Our package successfully selected subsets of gene expression data from the raw count data. Thus, TcellInflamedDetector can be beneficial for future cancer immunotherapy vaccine developers and researchers.