Introduction
In genome-wide association studies, many associations between single-nucleotide polymorphisms (SNPs) and phenotypes have been successfully discovered in many studies [1]. Despite the success of association studies, a large part of heritability remains unexplained as missing heritability [2]. Gene-gene interactions, rare variants, and structural variations are pointed to as causes of missing heritability.
For the detection of gene-gene interactions, the multifactor dimensionality reduction (MDR) method has been proposed by the reduction from genotype values of an SNP combination to a binary variable having a value of âhigh riskâ or âlow riskâ [3]. This method has been widely expanded for specific objectives, such as balanced accuracy for imbalanced data [4], generalized MDR for covariate adjustments and continuous phenotypes [5] for survival phenotypes [6, 7], and odds ratio-based MDR [8], etc. [9â14].
Among MDR expansions, fuzzy-MDR uses the fuzzy set theory for an adaptation of membership function for reflecting the uncertainty of âhigh riskâ or âlow risk,â and detection rate increases have been verified in many simulations [15]. Fuzzy-MDR has been expanded for covariate adjustments and continuous phenotypes [16] and maximum likelihood estimator as the membership function as empirical fuzzy MDR (EFMDR) [17].
In EFMDR, a maximum likelihood estimator for each genotype is a membership value of âhigh riskâ or âlow riskâ for the genotype. It has been proven that values of fuzzy balanced accuracy, based on maximum likelihood estimations, follow a chi-square distribution. Therefore, there is no need for cross-validation of p-value calculations. However, EFMDR is relatively slow, because it is implemented by R script only.
Methods and Results
To detect k-order interactions, MDR compares the balanced accuracy of all possible k-SNP combinations [3] and performs cross-validations. In fuzzy-MDR [15], fuzzy membership functions are used for reflecting the uncertainty of âhigh riskâ or âlow risk,â and validation of this uncertainty has been confirmed in various simulation experiments. In EFMDR [17], the maximum likelihood estimator of probabilities of âcaseâ or âcontrolâ is used as the relative membership degree of âhigh riskâ and âlow risk,â. In addition, it is proven that the fuzzy balanced accuracy of EFMDR follows a chi-square distribution and that the cross-validation scheme is omitted by this property [17]. Because of the omission of cross-validation and the simple membership function, EFMDR is faster than fuzzy-MDR, but it is still slow in detecting high-order interactions.
For fast execution of EFMDR written in R, such optimization methods as using vector calculations might be a good approach. Instead, we used the RCPP package [18] for the implementation of faster EFMDR. In EFMDR, for detecting the k-locus interaction of a p-SNP dataset,
( p k )
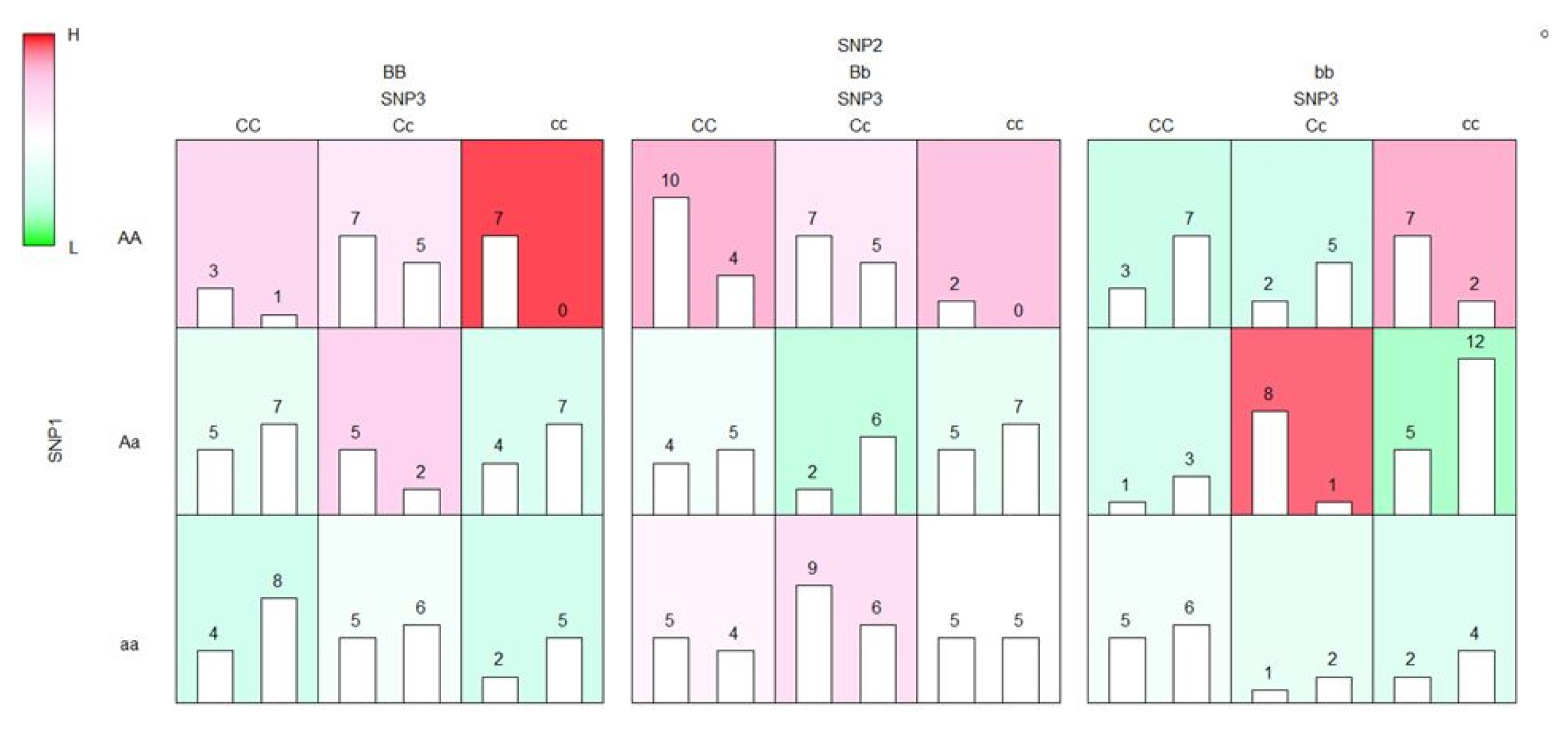
The results of the execution of EFMDR and EFMDR-Fast are in exactly the same formats. Hence, the results of EFMDR-Fast can be visualized easily using functions in EFMDR, as shown in Fig. 1.
We confirmed that the best SNP combinations of EFMDR and EFMDR-Fast are exactly the same. EFMDR-Fast is about 800 times faster than EFMDR. These comparisons were performed in R, version 3.5.1 on the Windows 10 platform with a 3.20 GHz CPU and 16 GB RAM. The program source codes and examples of EFMDR-Fast, written in R, and RCPP are available at http://statgen.snu.ac.kr/software/efmdr.