Effects of Somatic Mutations Are Associated with SNP in the Progression of Individual Acute Myeloid Leukemia Patient: The Two-Hit Theory Explains Inherited Predisposition to Pathogenesis
Article information
Abstract
This study evaluated the effects of somatic mutations and single nucleotide polymorphisms (SNPs) on disease progression and tried to verify the two-hit theory in cancer pathogenesis. To address this issue, SNP analysis was performed using the UCSC hg19 program in 10 acute myeloid leukemia patients (samples, G1 to G10), and somatic mutations were identified in the same tumor sample using SomaticSniper and VarScan2. SNPs in KRAS were detected in 4 out of 10 different individuals, and those of DNMT3A were detected in 5 of the same patient cohort. In 2 patients, both KRAS and DNMT3A were detected simultaneously. A somatic mutation in IDH2 was detected in these 2 patients. One of the patients had an additional mutation in FLT3, while the other patient had an NPM1 mutation. The patient with an FLT3 mutation relapsed shortly after attaining remission, while the other patient with the NPM1 mutation did not suffer a relapse. Our results indicate that SNPs with additional somatic mutations affect the prognosis of AML.
Introduction
Acute myeloid leukemia (AML) is the most common type of leukemia among hematologic malignancies. This disease is a clonal hematopoietic disease that is caused by either inherited or acquired genetic mutations. AML is known to be a molecularly and clinically heterogeneous disease [1]. In order to cure this disease, cytosine-arabinoside (Ara-C)-based chemotherapy is widely used. Two thirds of AML patients who receive Ara-C based chemotherapy regimen, will achieve complete remission (CR). However, more than 50% of patients suffer a relapse after attaining CR [2]. Eventually, a great majority of AML patients die from progressive disease after relapse by various genetic changes that will endow tumor cells with resistance against most commonly used anticancer agents. Therefore, it is prerequisite to identify prognostic markers that will help in deciding the suitable treatment modalities. A lot of studies have focused on finding somatic mutations in cancer cells by whole-exome sequencing (WES). However, these studies did not fully explain the mechanisms of how disease relapses.
In the initial development of cancer in humans, Knudson's two-hit hypothesis has provided important insights into the pathogenesis of tumors in autosomal dominant tumor predisposition syndromes [3]. With an analogy, we focused on the additional genetic changes that will predispose susceptible individuals with a pre-existing genetic abnormality to develop leukemia. To address this issue, germline single nucleotide polymorphism (SNP) analysis was performed using the UCSC hg19 program in 10 AML patients (samples, G1 to G10), and somatic mutations were identified in the same tumor sample using SomaticSniper and VarScan2. Our results suggest that SNPs with additional somatic mutations may directly affect the prognosis of AML and emphasize the need to validate in a larger patient cohort.
Methods
DNA sample preparation (normal and tumor samples)
Genomic DNA samples were prepared for WES. They were freshly obtained from AML patients' bone marrow at initial diagnosis. Subsequent bone marrow samples were aspirated if necessary according the disease status of the patients. AML blast tumor cells were isolated from bone marrow of each patient using Ficoll gradient separation. Matching patients' saliva samples were collected as germline controls. Each tumor and control DNA was extracted via standard protocols of DNA purification. DNA samples for validation were obtained from our frozen stock as needed.
Whole-exome sequencing
We prepared and sequenced exomes using the Solexa sequencing technology platform (HiSeq2000; Illumina, San Diego, CA, USA) following the manufacturer's instructions. We randomly sheared 3 µg of genomic DNA using the Covaris System to generate 150-bp inserts. The fragmented DNA was end-repaired using T4 DNA polymerase and Klenow polymerase, and Illumina paired-end adaptor oligonucleotides were ligated to the sticky ends. We analyzed the ligation mixture by electrophoresis on an agarose gel and sliced and purified fragments of 200.250 bp. The purified DNA library was hybridized with the SureSelect Human All Exons probes set (Agilent, Santa Clara, CA, USA) to capture 50 Mb of targeted exons following the manufacturer's instruction. We prepared the HiSeq2000 paired-end flowcell with the manufacturer's protocol using the captured exome library. Clusters of PCR colonies were then sequenced on the HiSeq2000 platform using recommended protocols from the manufacturer.
Read mapping and alignment and variant analysis
We recently developed an in-house analysis pipeline for cancer genome sequencing data. This pipeline essentially includes 1) mapping and alignment, 2) discovery of sequence nucleotide variations (SNVs) and insertions and deletions (indels), and 3) filtering and annotation of SNVs and indels. Briefly, once the raw sequence data were created, the output short reads were aligned to a reference genome (NCBI human genome assembly build 36) using UCSC hg19. Each alignment was assigned a mapping quality score by UCSC hg19, which is the Phred-scaled probability that the alignment is incorrect. The PCR duplicates were detected and removed by SAMTOOLS (http://samtools.sourceforge.net/). After alignment, we used GATK v1.4 to call SNVs for each chromosomal position. Filtering and annotation of SNVs and indels were performed using ANNOVAR (http://www.openbioinformatics.org/annovar/). We defined somatic mutation SNVs as those detected by both SomaticSniper and VarScan2 with a somatic p-value <0.05. We also set a minimum coverage of 5 in both normal and tumor samples. Conclusively, final somatic mutations were selected using the in-house filter.
Results and Discussion
Germline SNP analysis was done to detect germline heterozygous mutations that are carried by AML patients. To address this issue, germline SNP analysis was performed using the UCSC hg19 program in 10 AML patients (samples G1 to G10) (Table 1). We chose 18 major genes (TET2, ASXL1, DNMT3A, CEBPA, PHF6, WT1, TP53, EZH2, RUNX1, PTEN, FLT3, NPM1, HRAS, KRAS, NRAS, KIT, IDH1, and IDH2) to be analyzed in 10 samples (Table 2). Among these 18 genes, KRAS and DNMT3A seemed to play a critical role in the progression of AML, associated with the chemotherapy response. KRAS is a protein product of the normal KRAS gene, which performs an essential function in normal tissue signaling, and the mutation of a KRAS gene is an essential step in the development of many cancers [4]. DNMT3A is a key component of the epigenetic regulation of genes, as it catalyzes the addition of methyl groups to the cytosine residue of CpG dinucleotides [5]. However, variations only in KRAS and DNMT3A are not enough to explain the mechanism of AML progression, since additional somatic changes exist in AML patients.
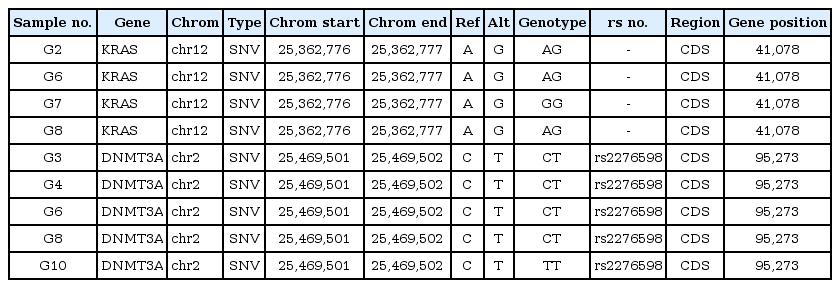
Germline SNPs (KRAS and DNMT3A) in 10 AML patients (samples, G1 to G10)
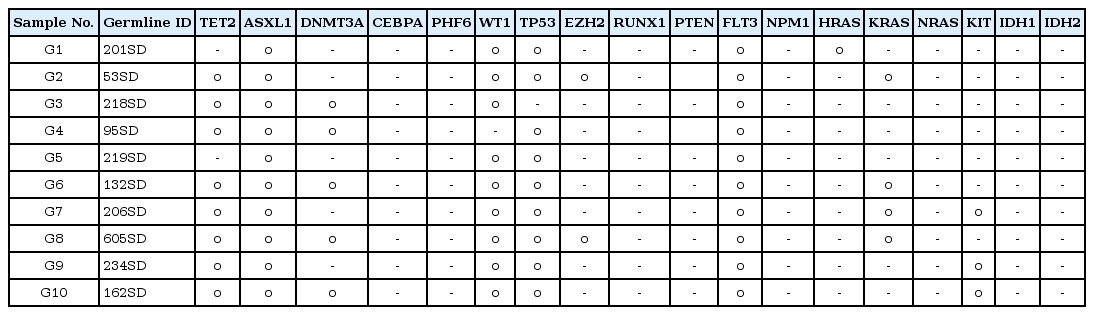
SNPs of KRAS were detected in 4 different individuals (G2, G6, G7, and G8), as shown in Table 1, while those of DNMT3A were detected in 5 individuals (G3, G4, G6, G8, and G10) among 10 AML patients (Table 1). Patients G6 and G8 showed SNPs in both KRAS and DNMT3A. We then searched for somatic mutations of each individual in order to prove the 'two-hit' theory. Table 3 shows the results of the somatic mutation analysis. SNPs of KRAS and DNMT3A were detected in both samples G6 and G8 (Table 3). However, they had different clinical courses after the first induction. They both attained a CR after the first induction; however, G6 relapsed within 390 days after attaining remission, while G8 did not relapse for 402 days. We suggested that an additional somatic mutation may have affected the clinical courses above. A somatic mutation in IDH2 was identified in both G6 and G8, while that of FLT3 and NPM1 was detected in G6 and G8, respectively. FLT3 mutation is known as a bad prognostic factor in AML [6]. NPM1 mutation, on the other hand, is usually known as a good prognostic factor in AML [7]. Therefore, we could speculate that SNPs (KRAS and DNMT3A) with additional somatic mutations (FLT3 and NPM1) affect the prognosis of AML differentially.
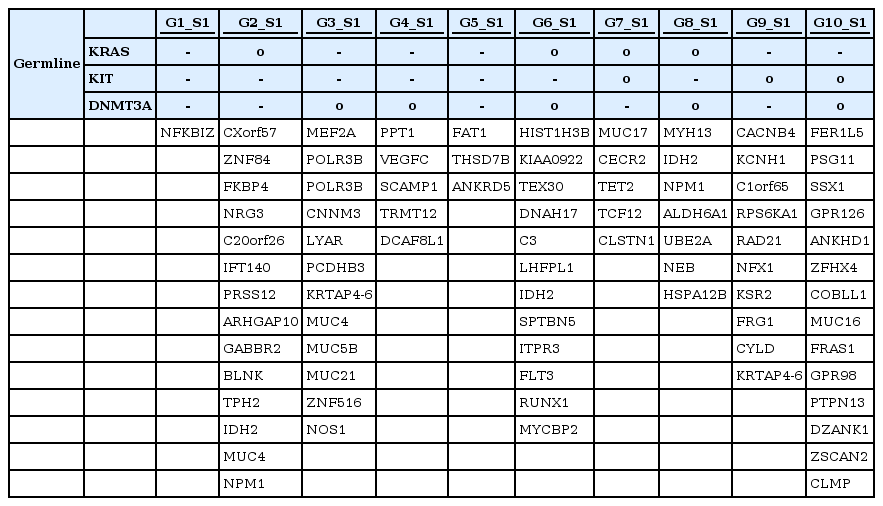
Somatic mutation calling results in 10 AML patients (samples, G1 to G10)
In addition, G2 and G7 shared the same SNPs in KRAS but their clinical courses showed a striking difference (Table 4). G7 had an additional germline SNP in KIT. This patient never attained CR despite intensive chemotherapy. As we searched further for somatic mutations in G7, a somatic mutation in TET2 was detected as a second hit. On the other hand, an NPM1 mutation was observed in G2, showing a divergent outcome compared with G7. G2 attained CR after the first induction of chemotherapy and did not relapse.
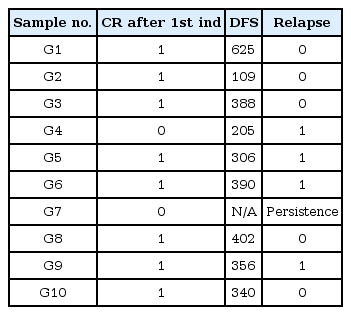
Clinical course of patients
We observed a divergent outcome in patients having the same SNPs but with different somatic mutations as a second hit. Somatic mutations that were differentially detected at the time of diagnosis strongly suggest clinical implications and warrant future study in a larger cohort to enhance our understanding of the mechanisms of disease progression in AML.
Acknowledgments
This study was supported by a grant of the National Project for Personalized Genomic Medicine, Ministry for Health & Welfare, Republic of Korea (A111218-GM05).