Analytical Tools and Databases for Metagenomics in the Next-Generation Sequencing Era
Article information
Abstract
Metagenomics has become one of the indispensable tools in microbial ecology for the last few decades, and a new revolution in metagenomic studies is now about to begin, with the help of recent advances of sequencing techniques. The massive data production and substantial cost reduction in next-generation sequencing have led to the rapid growth of metagenomic research both quantitatively and qualitatively. It is evident that metagenomics will be a standard tool for studying the diversity and function of microbes in the near future, as fingerprinting methods did previously. As the speed of data accumulation is accelerating, bioinformatic tools and associated databases for handling those datasets have become more urgent and necessary. To facilitate the bioinformatics analysis of metagenomic data, we review some recent tools and databases that are used widely in this field and give insights into the current challenges and future of metagenomics from a bioinformatics perspective.
Introduction
Metagenomics is defined as the study of the metagenome, which is total genomic DNA from environmental samples. Metagenomics has long been one of the major research tools in microbial ecology since the term was first used in 1998 [1]. Metagenomic information allows for a more in-depth understanding of the ecological role, metabolism, and evolutionary history of microbes in a given ecosystem by analyzing environmental DNA directly without prior cultivation. Metagenomics has made unprecedented contributions to microbial ecology; among them, one of the most outstanding discoveries of metagenomics is the first description of proteorhodopsin in marine bacteria [2]. Moreover, many hypotheses and questions in ecology and evolutionary biology have been tested and answered through metagenomic research. For example, Fuhrman et al. [3] tested a latitudinal gradient of biodiversity, which is one of the most widely recognized patterns in macroscopic taxa, on marine bacteria, and more recently, the taxonomic and functional distinction of bacteria in desert compared to other nondesert biomes was investigated through cross biome comparison using shotgun metagenomics [4]. Varied definitions converge into two main categories, targeted metagenomics and shotgun metagenomics, depending on their purpose and target materials. This review offers an overview of the tools and databases that are widely used in both targeted and shotgun metagenomics, in particular emphasizing on metagenome sequences generated by recent next-generation sequencing (NGS) technologies.
A Revolution of Sequencing Technologies and Current Challenges in Bioinformatics
The recent development of sequencing technologies has enabled us to assess much deeper layers of microbial communities by generating tons of nucleotide sequences at lower costs. NGS technologies have revolutionized the field of microbial ecology, as they have allowed researchers to reach the true level of diversity more closely through more in-depth sequencing. There are various applications using these NGS platforms, ranging from single-gene targeted sequencing to whole-genome sequencing and shotgun metagenome sequencing. Details about the principles and applications of each NGS technique have already been reviewed elsewhere [5]. The first NGS platform, pyrosequencer (GS20), generated just 20 Mb per run with a read length of 100 bp on average. A recent version of pyrosequencer (GS-FLX Titanium) has an advantage over other NGS techniques, especially 16S rRNA gene-based surveys, as it produces around ~500 Mb per run with a longer read length (400-500 bp), which is sufficient to cover partial hypervariable regions of 16S rRNA. Illumina sequencing produces many more reads with cheaper price that are more accurate (accuracy of >99%) than 454 platforms (accuracy of 98.93%) [6] but is somewhat limited in certain fields due to the relatively shorter read length (<100 bp) of early versions. However, Illumina became more popular, since it gradually improved readable lengths (i.e., 2 × 250 bp in MiSeq). More recently, Pacific Biosciences has released a new sequencing technology, PacBio RS, and Oxford Nanopore Technologies introduced GridION/MinION devices, both of which allow single-molecule sequencing with a much longer read length. However, further improvements are necessary for use in practice due to the high intrinsic errors (10% to 15% in PacBio and around 4% in GridION) [7].
Since the first launch of the NGS platform in 2006, novel sequencing technologies have been developed continuously and rapidly. As a result, massive sequence data produced by NGS technologies are accumulating at an unflagging rate. However, the computing power and development of algorithms needed to deal with the huge datasets efficiently are not keeping up with the speed of data production. For example, access to sequence data is still hampered by unsuitable data storage systems, such as short read archive (SRA), and many early papers were not accompanied by data deposition into public databases. Repositories should be big enough to be ready to allow the increasing volume of upcoming sequence data, and all data must be deposited in a standardized manner. In addition to the shortage of data storage space and confounding submission formats, the characteristics of the produced sequence data pose another problem in further processing. For instance, shorter read length, which is an inevitable limit of high-throughput sequencing, is a barrier for sequence assembly, and the relatively higher rates of sequencing errors compared with previous Sanger methods also make it hard to recover genuine sequences.
Targeted Metagenomics
Targeted metagenomics refers to a metagenomic approach that surveys genes or genomic regions of particular interest by targeted methods (activity- and sequence-driven studies) [8]. Sequence-driven targeted metagenomics is normally conducted by PCR-based directed sequencing of environmental genomic DNA. Ribosomal RNA genes (e.g., small subunit [SSU] and large subunit [LSU]) have been used as taxonomic marker genes for identifying microbial species. The characteristics of having both highly conserved and variable regions have allowed for accurate taxonomic identification and made it easier to design primers targeting the whole taxa of interest. Although taxonomic resolution of the 16S rRNA gene is not sufficient for delineating taxa at the species or strain level in some cases [9], the SSU of ribosomal RNA (16S rRNA) has been used widely as a standard marker gene in prokaryotes. For the last few decades, molecular methods to assess microbial diversity have focused on rRNA genes using probe-based approaches, such as fluorescent in situ hybridization and microarray, fingerprinting methods, and molecular cloning. Some of them are still in use, but in this review, we focus mainly on sequence data generated by recent NGS techniques.
Microbial community profiling using taxonomic marker genes (e.g., 16S rRNA gene) commonly uses an operational taxonomic unit (OTU)-based approach, as the sequence-based species definition in microbes is still vague and current public databases still do not reach the full extent of microbial diversity, despite the massive sequencing efforts. This OTU-based approach is now generally accepted in most microbial community studies based on environmental samples. Over the last few years, 454 pyrosequencing has been a major source of generating amplicon metagenomics data among NGS platforms due to its capability of producing a relatively longer read length. Therefore, bioinformatic analysis tools dealing with sequence data have been developed and tailored for pyrosequencing results. More detailed information about the algorithms and processes at each step can be found in several other reviews [7, 10]. In this part, we introduce recent tools and databases and provide brief explanations about how they work in the course of the analysis workflow (Table 1) [11-29].
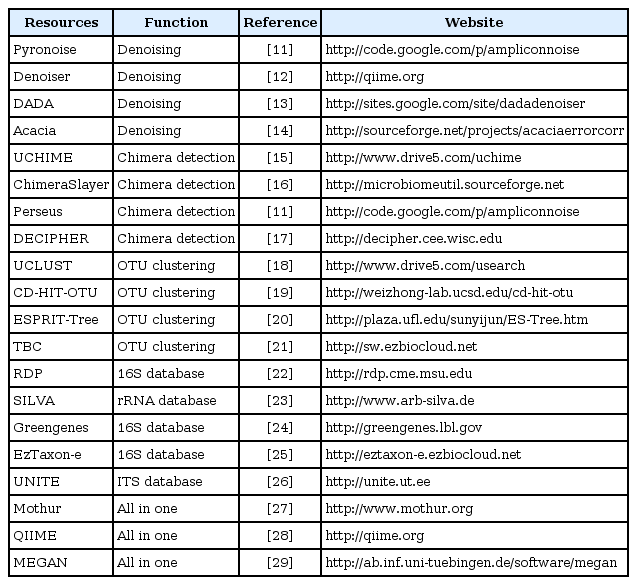
Bioinformatic resources for studying targeted metagenomics
Denoising
The first part of an analysis of NGS-generated data starts from filtering out 'noise' sequences. Most metagenomic studies based on single- or multiple-gene amplicons have used 454 pyrosequencing due to its advantage of producing longer read lengths, and currently available denoising algorithms have also been developed for that purpose. The denoising process per se does not remove actual sequences but keeps abundant information on erroneous sequences by retaining representative reads. Several denoising algorithms have been suggested so far. PyroNoise [11] implements a flowgram clustering method, and other denoising tools, such as Denoiser [12], DADA [13], and Acacia [14], use sequence abundance information on the denoising process. Similarly, single-linkage preclustering can be used before performing the formal OTU clustering to reduce 'noise' sequences generated by PCR and sequencing errors [30]. It first ranks sequences in order of decreasing abundance, and rarer sequences within a certain threshold are merged into the original abundant sequences.
Chimera Detection
Once denoising and additional quality control processes are completed, chimeric sequences should be removed from the dataset. Chimeras are artificial recombinants between two or more parental sequences, and they are normally formed when prematurely terminated fragments reanneal to other template DNA during PCR amplification [31]. These artificial molecules make it difficult to differentiate the original sequence from recombinants, resulting in overestimation of the level of microbial diversity in environmental samples [32]. Once chimeras are generated and sequenced, they need to be identified and removed from the dataset using bioinformatics tools. However, detecting chimeras is still challenging, as breakpoints can take place at any position more than once, and NGS platforms generate shorter lengths of sequences, making them hard to differentiate the source of parents with insufficient taxonomic information. Several elegant algorithms and tools have been suggested for preferentially identifying chimeric sequences in high-throughput datasets. These tools include UCHIME [15], ChimeraSlayer [16], Perseus [11], and Decipher [17]. All of these tools, except for ChimeraSlayer, use sequence frequency information to detect chimeras, assuming that chimeric sequences are less frequently represented in a given dataset than normally amplified sequences. There is no algorithm to detect chimeras perfectly, but to date, it has been known that UCHIME outperforms other algorithms, at least for short NGS reads [15]. Although there are still several limitations in detecting chimeric sequences that are formed by hybridization between closely related organisms, the tools listed above work well on SSUs and LSUs, but further validations are necessary for internal transcribed spacers and other functional genes.
OTU Clustering
The next step following chimera detection is OTU clustering, which is an essential process in community analysis, as it sorts sequences with the closest matches and then gives a taxonomic meaning to the clustered group. Sequencing errors, chimeras, and clustering algorithms have great influence on the quality of OTUs [33]. There are generally two ways that have been suggested for generating OTUs. One is alignment-based clustering, and the other is the alignment-free clustering method. Sequence alignment is done by either aligning query sequences against pre-aligned reference sequences [34] or using pairwise and multiple sequence alignments [35]. It is known that alignment quality has a significant impact on OTU clustering results [36]. Alignment quality varies, depending on gene loci and the parameters used, but sequence alignment incorporating secondary structure information generally improves OTU assignment, at least for 16S rRNA gene sequences [37]. The nearest alignment space termination (NAST) algorithm [34] has been used successfully in microbial ecology as a profile-based alignment tool, and SINA aligner [38], based on partial order alignment, and infernal [39], using consensus RNA secondary structure profiles, were recently introduced. However, there is currently no method or algorithm that does automatic alignment almost perfectly or up to the quality that can be achieved manually. Alternatively, alignment-free methods are more broadly used in picking OTUs. Commonly used alignment-free tools are UCLUST [18], CD-HIT [19], and ESPRIT-Tree [20]. UCLUST and CD-HIT implemented their own sorting processes in OTU clustering by sorting sequences in order of decreasing abundance (i.e., UCLUST) or decreasing length (i.e., CD-HIT), and ESPRIT-Tree uses an average linkage-based hierarchical clustering algorithm. There are some other tools that implement a 'homopolymer collapse option' for minimizing homopolymer errors in 454 reads: CLOTU [40], SCATA (http://scata.mykopat.slu.se), CrunchCluster [41], and TBC [21]. However, these tools are currently applicable only to 454 reads, and there is a fundamental limitation of not being able to detect 'real' sequence differences appearing in homopolymer regions. SCATA has suggested another OTU picking approach by using both reference sequences and query sequences together in the clustering process, and then the OTU is assigned to a taxonomic identity of the reference sequence if the reference sequence belongs to the cluster.
A taxonomy-dependent approach was recently proposed as an alternative to OTU clustering approaches [42]. This method has two major advantages over OTU clustering. First, direct taxonomic assignment to each query sequence is more tolerant to sequencing errors than the OTU picking process, as the assignment process is less affected by mismatches or insertion/deletion errors, while sequencing errors are known to generate many spurious OTUs. It also helps prevent a massive loss of erroneous but still taxonomically meaningful sequences. Second, it enables researchers to perform a more standardized community analysis, based on a single assignment rule. There is no truly 'universal' primer set, and the difference in primer sets makes it difficult to compare datasets, as the different primer sets amplify differing variable regions within 16S rRNA genes and catch only a partial body of the whole 'true' community. On the other hand, a taxonomy-supervised method can circumvent the problem by directly assigning sequences to the closest relatives using full-length reference sequences. This approach also facilitates cross-comparison and meta-analysis across versatile microbial community studies. A caveat is the low taxa coverage of current marker gene databases, which offsets the advantage of this approach and hinders the application in practice by failing to find the closest matches to known members, due primarily to its lack of close relatives in a given database. Another limitation is the relatively lower assignment accuracy and precision of short NGS reads compared to those of full-length sequences. It is generally known that even longer 454 Titanium reads are able to be identifiable with high confidence scores at best to the genus level [43], even genus-level classification is doubted [44]. However, this problem will be soon overcome as more high-quality sequences are added to the databases. For example, EzTaxon-e benefits from using artificially defined species names, which are defined based on combinatorial evaluation using both phylogeny and pairwise similarity between unclassified sequences on all taxonomic levels. It endows taxonomically more meaningful information to those 'unclassified' sequences rather than remaining unknown or environmental sequences. Moreover, recent updates have slightly improved the database coverage by adding error-free 454 reads that are selected using abundance information (http://eztaxon-e.ezbiocloud.net/).
16S Databases and Taxonomic Classification
Identifying each individual sequence is of great importance in microbial community analysis, as taxonomic information gives us access to basic information of its traits, such as physiology, epidemiology, and evolutionary history, and it allows for indirect inference of their ecological roles in a given environment. There are several methods that have been suggested for assigning microbial taxonomy with high-throughput sequence data. BLAST [45] is one of the most widely used algorithms when classifying sequences. The top BLAST hits by searching against reference databases are generally used for taxonomic identification of query sequences. There are several well-curated public SSU databases, such as RDP [22], SILVA [23], Greengenes [24], and EzTaxon-e [25]. However, the top BLAST hit does not always give the correct taxonomy result, especially in shorter reads. Alternatively, Greengenes and EzTaxon-e use BLAST in conjunction with global alignment, and the post processing of a BLAST-aligned output has been suggested to improve assignment accuracy using the lowest common ancestor (LCA) algorithm in MEGAN [46] and the optimal consensus method (F-measure) in TANGO [47]. The probabilistic approach also gives a similar level of accuracy but is much faster than a BLAST search. The naïve Bayesian algorithm [48], implemented in RDP, uses 8-mer word-matching for training datasets and provides assignment results with bootstrapped confidence estimates. Phylogenetic placement method is a recently suggested approach that places query sequences into a phylogenetic guide tree on the basis of various evolutionary models. This approach is particularly useful in a case where there are no close relatives to the query sequence in databases, due primarily to the low DB coverage, which is the case with microbial eukaryotes. Tools using phylogenetic methods in taxonomic assignment include the evolutionary placement algorithm (EPA) [49], pplacer [50], and SEPP [51], and these algorithms were recently incorporated into QIIME [28] and AMPHORA2 [52].
Shotgun Metagenomics
Until the arrival of NGS technologies in this field in 2006, shotgun libraries had been constructed using circular vectors, each of which had an insert derived from a metagenomic DNA fragment [53]. Sequencing strategies designed either to completely assemble the sequence of each long-insert vector (tens to thousands of kilobases) or to generate a paired-end read from small inserts (around 1-2 kilobases) were applied to those vector libraries. The former strategy has a unique strength, in that it is relatively easy to generate contigs orders of magnitude longer than the read length when assembling the reads from homogeneous templates [2]. However, such a strategy is not scalable to analyze a large number of clones, as the tremendous amount of reads per clone needs to be screened. The latter strategy, a genuine random shotgun approach, took essentially the same approach with the NGS-based metagenome assembly, which enables analysis of a large number of clones but requires high coverage in order to be successful [54, 55]. Regardless of the strategies taken, vector library construction and subsequent sequencing are time-consuming and cost-ineffective, and only a few organizations can lead such efforts.
NGS technologies have proven their utility in shotgun metagenomics since their earliest application appeared in 2006 with studies using 454 pyrosequencing data. After all, technologies like 454 and Illumina sequencing have become routinely applied for shotgun metagenomics, accompanying changes in the trends of the field as well as the properties of the data themselves. As sequencing cost continues to decrease, more researchers outside the big sequencing centers have started to participate in metagenomic studies, resulting in higher individuality and diversity of metagenome data. Bioinformatics for metagenome analysis has progressed, along with the explosion of data, both by extension from the tools and databases previously developed in genomics and the adoption of methodologies used by ecologists. Less than a decade has passed since metagenomic shotgun sequencing took its place as general practice for biologists, and it is fair to say that bioinformatics for shotgun metagenomics is at a premature stage. In this section, we aim to summarize the methods that seem to be the 'current' standard, the most widely used, in the analysis of shotgun metagenomic data (Fig. 1).
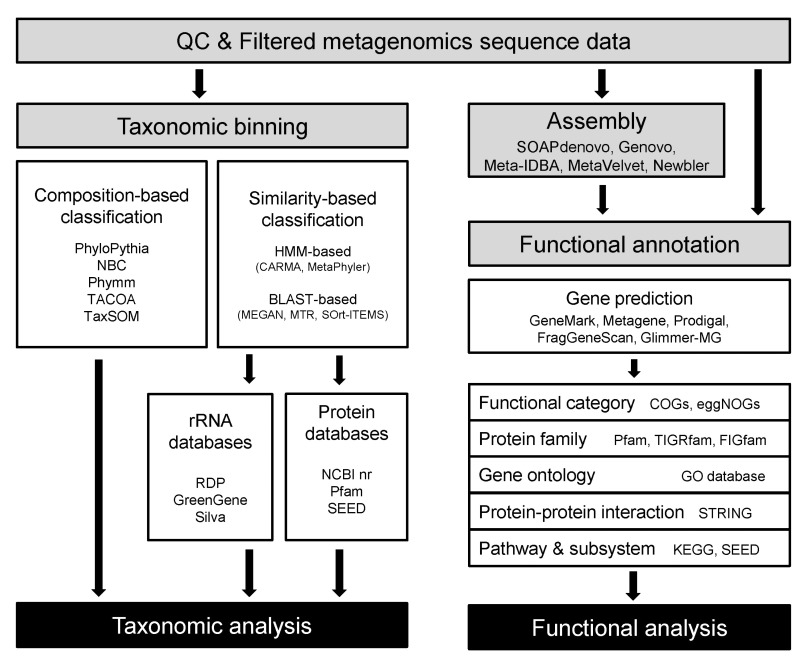
Overall workflow and bioinformatics tools for shotgun metagenomic analysis. HMM, hidden Markov model; KEGG, Kyoto encyclopedia of genes and genomes; STRING, Search Tool for the Retrieval of Interacting Genes/Proteins.
Assembly
Assembly is the process of merging overlapped short reads into larger contiguous sequences (contigs). Current sequencing technologies normally break genomic DNA into pieces, and original genomic sequences need to be recovered from fragmented reads by an assembly process. In metagenomics for retrieving coding regions, it is necessary to make it easier to do functional annotations, as a more accurate classification/annotation is possible in longer sequence information. Most assemblers have been developed for the single genome or clonal populations with high coverage. There are currently few metagenomic assemblers, as metagenomic reads are more complex, owing to nonclonal heterogeneous reads resulting from multiple strains differing only by partial regions or rearrangements, lower or uneven coverage across genomes, sharing of repetitive sequences among closely related species, and lateral gene transfer sharing similar entities between distantly related organisms.
At an early stage of metagenomic studies, reference mapping approaches (e.g., Newbler, AMOS, MIRA) were applied to the metagenome assembly process. However, with the low coverage and complexity of megenomic reads, de novo assemblers were preferred to reference mapping, based on the advantage of feasibility for dealing with the aforementioned problems. Starting from initial assemblers, such as SOAPdenovo [56], there are now several improved assemblers available: are Genovo [57], Meta-IDBA [58], Meta-Velvet [59], MAP [60], and Ray Meta [61]. Metagenome-specialized assemblers have to distinguish reads from different species. MetaVelvet and Meta-IDBA resolve this issue by partitioning the de brujin graph based on k-mer coverage and separately assemble each subgraph. Ray Meta does not decompose the de brujin graph; instead, it uses a heuristics-guided graph traversal approach to find the optimal assembly. Meta-IDBA, MetaVelvet, and Ray Meta were developed to perform well on short reads (e.g., Illumina sequencing). On the other hand, other tools, like Genovo and MAP, were developed for longer reads (e.g., 454 sequencing). The outputs from various assemblers can be used to generate scaffolds using Bambus2 by avoiding misjoins between distantly related organisms by detecting repeats and genomic variants [62].
Binning and Taxonomic Classification
Taxonomic classification of reads, which is called binning, is the most basic step in the characterization of microbial communities by metagenome sequencing. However, taxonomic binning of metagenome shotgun sequences is a challenging task for researchers, especially when working with short reads derived from NGS. There are several reasons that make binning a nontrivial job.
One of the reasons is that NGS technologies generally produce short reads. When applying shotgun sequencing to complex microbial communities found in soil, seawater, freshwater, and the gastrointestinal tract, the sequencing coverage does not reach the level required to make the assembly practically useful, even by current deep sequencing methods. The majority of reads remains unassembled. As a result, the majority of reads is taxonomically classified by the information in their short length. Short length presents a number of drawbacks in binning. Homology search-based methods suffer from a lack of alignment confidence and difficulties in predicting protein sequences from partial genes. Phylogenetic methods suffer from a lack of resolution due to insufficient phylogenetic information. Another challenge is the size of data that have to be processed during the binning process. Data size here refers to both the reads obtained from metagenomes and the sequences in the reference database. The novelty of microbes in the environmental samples also hampers binning, as they are not represented by any sequence in the reference database. One last challenging thing for researchers is the diversity of binning tools. There are dozens of choices, differing in both logical and practical aspects.
A straightforward approach for taxonomic classification of metagenome shotgun reads is searching for a similar sequence in a collection of known sequences that carries the taxonomic identity of each sequence. Similarity-based methods vary in at least three dimensions. 1) Choice of reference database: the search space could be restricted to a particular marker gene (e.g., SSU rRNA for prokaryotes) or a small number of selected protein families or could be as general as the entire NCBI nr database. 2) Search algorithm: from BLASTN (in case of using rRNA gene marker), BLASTX (protein database), and BLASTP (protein database after gene prediction) to a hidden Markov model (HMM)-based homology search (searching for protein families). 3) Taxonomic assignment from the hit information: from simply transferring the identity of the best hit to the use of LCA, modified LCA, or more complicated phylogenetic inferences. Among the popularly used tools, MEGAN uses the NCBI nr database for searching and LCA algorithms for assignment [29]. MTR and SOrt-ITEMS modified MEGAN's LCA algorithm by use of taxonomic information shared by hits (MTR) or by performing a reciprocal BLAST hit to reduce false positive hits (SOrt-ITEMS) [63, 64]. MG-RAST exploits both rRNA gene sequences and protein-coding sequences [65]. For the rRNA gene sequences, MG-RAST follows the RDP pipeline [22], while for protein-coding genes, it first predicts the protein coding sequences and then performs a BLASTX search against a number of databases, including SEED [66], and extracts taxonomic information from SEED hits. CARMA uses an algorithm similar to LCA and offers two ways for searching the database of known protein sequences: the NCBI nr database searched by BLASTX and the Pfam database searched by HMMER3 [67]. MetaPhyler is a power-up version of AMPHORA and uses 31 marker genes to reduce the search space [68, 69].
Composition-based classification of DNA sequences has been proven to be very useful, although it does not appear as intuitive as similarity-based methods. Composition-based methods exploit the uniqueness of base composition (from single to oligonucleotide levels) found across the genomes of different taxonomic entities. In many cases, these methods implement machine-learning algorithms. Ultimately, all of these methods use the taxonomic information of the reference database to assign a taxonomic identity to the reads. However, they can be divided into supervised and unsupervised methods, based on their dependence on the reference training set during the initial learning procedure. The most popular tools using supervised learning are PhyloPythia, NBC, and Phymm. Starting from the publicly available microbial genome sequences, NBC trains a naïve Bayes classifier based on the N-mer frequency profiles of each genome [70], Phymm builds an interpolated Markov Model using variable-length oligonucleotides typically found in taxa [71], and PhyloPythia trains a support vector machine classifier based on variable-length oligonucleotide composition [72]. Among the above, NBC and Phymm are suitable for classifying short reads generated from NGS sequencers. The popular tools employing unsupervised learning are TACOA and TaxSOM. TACOA introduced a kernelized k-nearest neighbor approach to cluster the reads [73], while TaxSOM uses batch-learning self-organizing maps (BLSOMs) and growing SOMs (GSOMs) to generate the clusters of related reads [74]. Both TaxSOM and TACOA are not suitable for unassembled short NGS reads.
There are tools that combine composition-based approaches and similarity-based approaches together to gain accuracy and discard fewer reads. PhymmBL linearly combines the BLASTN score and Phymm score and thereby gains more power to discriminate between similar BLAST hits [71]. RITA also combines a BLAST search with a composition-based NBC; however, unlike PhymmBL, RITA puts more weight on the BLAST result [75]. Both approaches are reported to perform well, even with short reads, but as they involve a BLAST search, they consume much time. There is a third type of taxonomic binning method recently implemented in the tool MetaPhlAn, which uses clade-specific marker genes [76]. From the database of microbial genomes, MetaPhlAn precalculated 400,141 genes that are most representative of each taxonomic unit. In theory, detection of the reads that match these markers can classify the members at the species level. MetaPhlAn uses BLASTN search to compare the reads against the set of marker genes. As the search space is markedly reduced from general sequence databases used in other approaches, MetaPhlAn exhibits unusually high speed.
Functional Annotation and Metabolic Re-Construction
Functional assignments and pathway reconstructions are ultimate steps of shotgun metagenomics that allow the characterization of the functional potential of uncultivated microbes or microbial communities under investigation. Connecting the sequences of metagenomic data to specific functions in the environments can be performed by using web-based workflows offered by useful portals without access to high-performance computers. These online metagenome annotation services, like IMG/M [77], METAREP [78], CAMERA [79], and MG-RAST [80], provide platforms for gene prediction, assignment of functional categories, protein families and gene ontologies, and inference of protein interactions and metabolic pathways represented in the metagenomic data (Table 2). While an installable workflow like MEGAN4 [29] also exists, MEGAN4 does not provide gene predictions or homology-based or profile-based annotation analyses. Instead, the pipeline uses ready-made annotation data derived from BLASTX or BLASTP against the NCBI nr database.
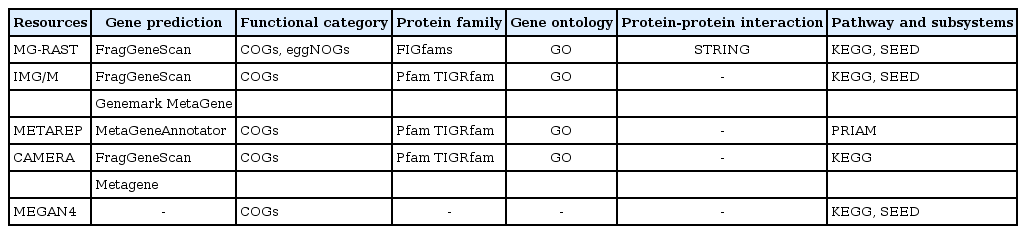
Comparison of five major resources for metagenomic functional annotation
Gene finding or gene prediction is a fundamental step for annotation. Classical gene finders that have been developed for a single genome are unsuitable for metagenomic analysis, because metagenome data are made up of a mixture of sequences from different organisms and often comprise mainly short assemblies and unassembled reads. Moreover, the high error rate of NGS can lead to frameshifts and make gene prediction more difficult. For this reason, several dedicated gene prediction programs have been developed, like Meta-Gene [81], MetaGeneAnnotator [82], Orphelia [83], FragGeneScan [84], Glimmer-MG [85], and MetaGenemark [86]. These programs incorporate different models for gene prediction, such as machine learning algorithms [87], HMMs [88], and di-codon usages [89]. MetaGeneMark (GeneMark) uses codon usage-incorporated HMM; Prodigal incorporates a machine learning algorithm [90]; MetaGene incorporates di-codon usage [91]; FragGeneScan uses a sequencing error model and codon usage-incorporated HMM; MetaGeneAnnotator uses a machine learning algorithm and di-codon usage information; Glimmer-MG uses an interpolated Markov model [92]; and Orphelia uses a codon usage-incorporated machine learning algorithm.
Functional analysis of metagenome generally uses a homology-based approach and involves a BLAST [45] search against a database, integrating several individual databases curated for specific analysis [93]. For functional category annotation, COGs [94] and eggnogs [95] databases are used. COGs, one of the widely used gene categories, was constructed from 66 genomes. Because COGs has a relatively small size of functional categories appropriate for determining well-known genes and because the database has not been updated for a long time, COGs shows low sensitivity for recently discovered genes. eggNOGs, created in 2011 and used by the portal service MG-RAST, shows higher sensitivity than COGs, because it is constructed based on the pre-annotation of orthologous groups from 1,133 genomes using COGs and KOGs. A BLAST-based approach is still widely used in functional category annotation, but it is hampered by its computational complexity and lack of homologous sequences in reference databases [93].
For a protein family analysis, the resources in the Pfam [96] and TIGRfam [97] databases are applied through the use of a HMM-based algorithm, profile (HMM profile), and search tool (HMMER; [98]). Similarity search results are transferred to annotations, based on the best hit's information. The Pfam and TIGRfam databases are resources consisting of curated multiple alignments and HMMs generated from the Sanger Institute and J. Craig Venter Institute, respectively. A previous study reported that more protein family predictions were available using Pfam, because the Pfam database contains a higher number of protein families (14,831 protein families in Pfam 27.0 release) than TIGRfam database (4,284 protein families in TIGRfam 13.0 release) [93]. While IGM/M, METAREP, and CAMERA use HMM-based databases, such as Pfam and TIGRfam, MG-RAST uses an HMM-independent database, namely FIGfams [99]. FIGfams are sets of protein sequences and alignments that are similar along their full length and are believed to implement the same function generated from National Microbial Pathogen Data Resource (NMPDR). Gene ontology is assigned using the gene ontology (GO) database in most publically available pipelines. GO analysis was available in version 3 of MEGAN, but it was replaced by two functional methods, based on the SEED classification and Kyoto Encyclopedia of Genes and Genomes (KEGG) in MEGAN4. In addition to protein family analysis, MG-RAST offers information on putative protein-protein interactions by applying EMBL's Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) tool [100].
Metabolic pathway reconstruction is generally performed using the KEGG database [101]. While IMG/M, CAMERA, and MG-RAST use the KEGG database and KEGG graphs, METAREP uses PRIAM [102]. PRIAM is a method for automated enzyme detection, based on the available ENZYME database [103], and provides KEGG graphs for visualization. Other pathway databases and tools, like MetaCyc [104] and MetaPath [105], are also used to annotate functional roles for metagenomic data but have not yet been integrated into online portal services for metagenomic analysis. An alternative to the methods mentioned above for inferring metabolic pathways is 'subsystem' in SEED [66]. Subsystems represent the collection of functional roles that constitute similar or related forms of complex structural and metabolic pathways, such as glyoxylate bypass, sulfur oxidation, and ribosomal protein paralogs. The subsystem annotation based on SEED is currently implemented in IMG/M, MG-RAST, and MEGAN4.
Future of Metagenomics
Early metagenomic studies focused mainly on investigating less complex ecosystems and specific targets in a given environment due to high sequencing costs, ecosystem complexity, and lack of high-performance computing and bioinformatics tools. Much work has been done on testing new molecular techniques and tools and discovering novel enzymes and taxonomic lineages rather than testing many scientific hypotheses. This is why many previous and even current studies are not sufficiently replicated in experimental design. Of course, it is possible to compare the difference between metagenomic studies, which are not replicated [106]. However, it enables us to obtain statistically meaningful and biologically more relevant conclusions if larger sample sizes with more replicates are provided at a deeper layer of the sequencing regimen.
For inferring statistically meaningful differences between metagenome samples and exploring metabolic/taxonomic diversity in a given sample, metadata should be provided and standardized according to a proper rule. More standardized formats for describing marker genes, genomes, and metagenome datasets were recently suggested in minimum information about any (x) sequence checklists (MIxS) [107]. This standardized format makes it possible to do many statistical analyses and meta-analyses across metagenomic studies by providing more standardized contextual information about the environment sampled as well as experimental and sequencing information. Centralizing contextual information will become more common in future metagenomic studies.
Aside from experimental design and contextual data, metagenomic data have inherent limitations that must be overcome in the future. Metagenomic reads commonly show a relatively low genomic coverage compared to that of a single genome, and the short length of sequencing reads makes only fragmented information by the incomplete assembly and annotation processes accessible. Initiatives are already under way for filling the gap between metagenomic reads by doing co-assembly with single-cell genomics [108] and joint analysis between multiple metagenomes simultaneously [109], on the assumption that the same species must exist in different samples and that the co-occurrence helps extract shared information. The ultimate goal of metagenomics is a comprehensive understanding of our ecosystem. In the near future, metagenomics will be one of the essential parts of viewing our ecosystem through integration with other '-omics' approaches such as metatranscriptomics and metaproteomics.
Conclusion
Over the last few years, metagenomics has accelerated the understanding of microbial ecology and evolution, thanks to the technical advances of sequencing platforms. Metagenomics itself is not a panacea that is able to answer all unresolved questions and uncover the 'dark matter' of our knowledge in microbial diversity and function studies, but it is undoubtedly an excellent tool, giving insight into completing the jigsaw puzzles of our understanding of the surrounding biosphere by exploring both 'who is out there' and 'what they do' in nature. It is true that studying metagenomics is still recognized by ordinary microbial ecologists as a tool that is difficult to tackle due to its complexity and the enormous amount of sequence data that needs to be dealt with. However, it is not surprising that we will witness 'doing metagenomics' in the future as if we routinely do PCR and gel electrophoresis in our laboratory. Current development of tools and databases for metagenomic studies is in its infancy, and there are still many challenges to overcome. It will not take long to see an explosive growth of metagenomics with a revolution of metagenomic bioinformatics.
Acknowledgments
This work was supported by the National Research Foundation grant (2013-035122) through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology of the Republic of Korea.