Introduction
High-throughput genomic technologies (HGTs), including next-generation DNA sequencing (NGS), microarray, and serial analysis of gene expression (SAGE), have become effective tools for cancer genomics through various applications. Especially, the applications of NGS include whole-genome, exome, and transcriptome approaches for searching for a wide range of cancer-specific genomic alterations, such as point mutations, insertions, and deletions; copy number changes; and rearrangements, leading to the development of cancer [1].
One of the hurdles in HGT-based cancer genomics is to identify causative mutations or genes out of many candidates from the analysis. A useful approach is to refer to known cancer genes and associated information [2]. The list of known cancer genes can be used to determine candidates of cancer driver mutations, while cancer gene-related information, such as gene expression, protein-protein interaction, and pathway information, can be useful for scoring novel candidates.
Some useful cancer genomic databases for this purpose exist, including Cancer Gene Census (CGC) [3] and Catalogue of Somatic Mutations in Cancer (COSMIC) [4], which have been constructed and managed by the Cancer Genome Project of the Welcome Trust Sanger Institute; and Cancer Gene Index (CGI, http://ncicb.nci.nih.gov/NCICB/projects/cgdcp), The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov), and Cancer Genome Anatomy Project (CGAP, http://cgap.nci.nih.gov), which are maintained by the National Cancer Institute of the U. S. National Institute of Health and National Human Genome Research Institute. Among them, COSMIC and CGC are the two most commonly used resources among researchers when checking reported cancer driver mutations. The COSMIC database stores information on somatic mutations and associated information extracted from the scientific literature, while the CGC database is a catalog of cancer genes with manually screened somatic mutations extracted from the literature. These cancer genes are annotated with information concerning chromosomal location, tumor types in which mutations are found, classes of mutations that contribute to oncogenesis, and other genetic properties. Also, CGI is known to be created through an automated linguistic text analysis of millions of MEDLINE abstracts, with manual validation and annotation of the extracted data by expert curators. It is a high-quality data resource consisting of genes that have been experimentally associated with human cancer diseases and/or pharmacological compounds, the evidence of these associations, and relevant annotations on the data. Thus, it can also be a valuable resource to annotate mutation data from cancer genomics studies.
Currently, few specialized tools exist for an automated analysis of a long gene list from HGT-based cancer genomics. While previously described cancer genomic information databases can be useful, they are not effective in processing results from NGS-based cancer genomic studies. Meanwhile, there are many functional annotation systems to analyze gene lists derived from high-throughput microarray experiments, including DAVID [5], GoMiner [6], GOstat [7], Onto-express [8], and gene set enrichment analysis (GSEA) [9]. DAVID is one of the most popular general purpose functional annotation systems. It provides various effective analysis tools and is also applicable in the analysis of gene lists from cancer genomics research. However, DAVID is not optimized for processing results from HGT-based cancer genomics data, especially for NGS-based cancer genomics downstream data; therefore, a more specialized cancer gene annotation system is needed.
Here, we introduce a web-accessible cancer genome annotation system, named CaGe, to provide users with information on cancer genes, mutations, pathways, and associated annotations, based on several cancer gene databases composed of reported cancer-causing genes and associated cancer pathways. For a given gene list, CaGe searches cancer gene databases with converted standard gene symbols, analyzes the biological pathways, and provides various cancer gene-related annotations through intuitive web user interfaces. It also serves additional functions, including processing various input types and formats, managing jobs for user submitted tasks, and browsing for cancer genes and pathways with various useful hyperlinks between the annotations and the external public annotation databases. We hope CaGe will be useful in identifying cancer-causing mutations and genes in HGT-based cancer genomics.
Methods
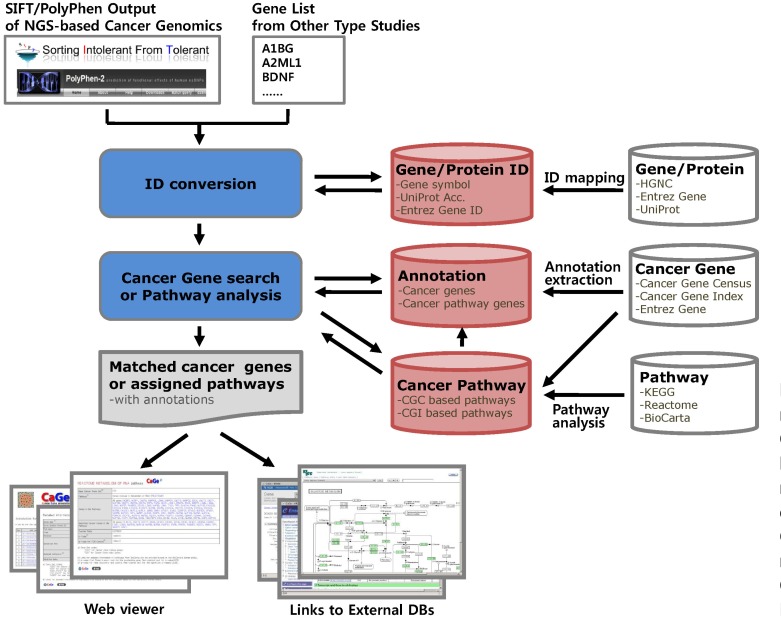
The goal of this study was to provide researchers with a tool for classifying their candidate genes from HGT-based cancer genome studies into previously reported or novel categories of cancer genes, while providing insight into underlying carcinogenic mechanisms through a pathway analysis. To implement the cancer gene annotation function of CaGe, we constructed reported cancer gene and cancer annotation databases from public cancer genomic databases and cancer pathway-gene databases by pathway analysis with reported cancer gene sets and canonical pathways. We also constructed a gene ID database to allow various input formats for the input of gene lists and a gene functional annotation database to provide users with functional clues for the annotated candidate genes. Then, we developed a core retrieval program and web interfaces for the main functions, which include cancer gene annotation, cancer pathway annotation, cancer gene browsing, and cancer pathway browsing. The workflow for the database construction and data processing in CaGe is summarized in Fig. 1, and the cancer gene annotation page of the CaGe web interface is shown in Fig. 2.
Cancer gene and annotation data
To construct the reported cancer gene database, we used gene sets from the CGC database (released on Dec, 2010) and CGI database (downloaded on Feb, 2011). The cancer pathways for the cancer pathway gene database construction were assigned based on statistical significance from one-tailed Fisher's exact test for overlapping genes between reported cancer gene sets and canonical pathways from public pathway databases, including KEGG (Release 57.0), BioCarta, and Reactome (downloaded on Feb, 2011).
We also created a gene ID database to convert various input identifiers into standard gene symbols with HUGO Gene Nomenclature Committee (HGNC) (downloaded on Feb, 2011) data for the standard gene symbols and with Entrez Gene (downloaded on Feb, 2011) and UniProt (Release 2011_03) data for the gene IDs, protein IDs, and functional annotations.
Input types
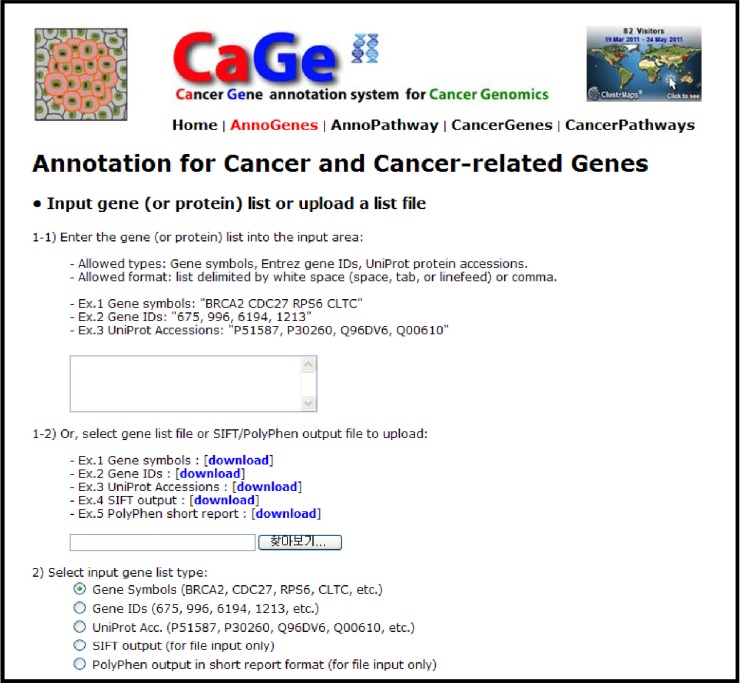
Available input types include a list of gene symbols, Entrez gene IDs, or UniProt accessions and output files from prediction tools, such as Sorting Intolerant From Tolerant (SIFT) [10] and PolyPhen [11], which evaluate functional effects of mutations on proteins. CaGe parses the output of those tools to extract the list of genes that have somatic mutations evaluated as functionally damaging, classify them into previously reported and novel candidate cancer genes, and conduct pathway analyses to give insights into underlying carcinogenic mechanisms. Thus, CaGe offers straightforward data processing from HGT-based data without additional data conversion.
Cancer gene and pathway annotation workflow
After acquiring user input, CaGe converts various input gene IDs into standard gene symbols, finds known cancer genes and pathways, links various cancer-related annotations to matched genes, and outputs them in the form of tables or text files through the web interface (Fig. 3). Another function of CaGe is to identify over-represented cancer-related or other biological pathways from the input gene list by performing one-tailed Fisher's exact test.
When processing more than one annotation job, users can manage their annotation tasks through the CaGe interface, and access to their results is maintained by the internal job database of CaGe until the jobs are deleted by the user or by a scheduled cleaning process. The IP addresses of client computers are used for secured job management without a logon process. Completed annotation jobs are listed on the job table, and the annotated results can be shown selectively by the user on the annotation result page or can be downloaded as tab-delimited text files for further analyses. The annotation on the results page has many useful links to gene and cancer-related information in the CaGe database or external public databases. In addition to the cancer gene and pathway annotation function, CaGe provides a function to browse cancer genes and pathways so that users can search cancer-related annotations without an input list. The search flows of cancer genes and pathways are connected to each other by crosslinks in the cancer gene information page or pathway information page.
Results and Discussion
Database and web interface
Based on the reported cancer-related gene sets from CGC and CGI, a cancer gene database and a cancer pathway gene database were constructed by the described methods. In total, 436 CGC-originated and 7181 CGI-originated cancer genes were prepared for the reported cancer gene annotation. Additionally, 5,790 CGC-based and 6,744 CGI-based cancer pathway genes were assigned for the annotation of unreported but potential candidate genes (Table 1). The gene ID database for gene symbol conversion, based on HGNC, and the functional annotation database, based on Entrez Gene and UniProt database flat files, were constructed. The types of annotations provided by CaGe are summarized in Table 2.
The main window of the CaGe web interface is shown in Fig. 2 for the cancer gene annotation process. Main menus, located in the upper part of the main window, are linked to the four main functions and home page of the system: 1) home page, 2) cancer gene annotation function, 3) cancer pathway annotation function, 4) cancer gene browsing function, and 5) cancer pathway browsing function. Detailed usages for CaGe are described in the user's guide at http://mgrc.kribb.re.kr/cage/include/cageUserGuide.pdf.
System information
CaGe was developed using PHP, R, and python languages; MySQL for database management; and Apache for the web server and is operated on a Linux platform with 8-core Intel Xeon CPUs (2.33 GHz) and 24 GB of main memory.
Performance evaluation with small cell lung cancer mutation data
To assess the capability of CaGe, we annotated genes from candidate mutations for a small cell lung cancer genome (SCLC) [12]. We had 59 genes with predicted functionally damaging mutations after applying 22,910 mutations to the PolyPhen and could annotate 22 previously reported known cancer genes successfully by applying the PolyPhen output for CaGe. Known cancer genes included RB1, which was mentioned as an SCLC-related gene in Pleasance's work; DST and ETS2, which were previously reported as SCLC-related genes but not mentioned in Pleasance's work; and 3 more genes (PDGFC, IL16, and AGTR2), which are known as lung cancer-related genes (Supplementary Table 1). The other 16 genes are known to be related to other cancer types, suggesting that they might be important in the carcinogenesis of SCLC as well. From the pathway analysis, we identified 12 known cancer genes and 11 genes in cancer-related pathways (Supplementary Table 2). Those 11 genes might also be important in the carcinogenesis of SCLC. Thus, we conclude that CaGe can annotate cancer genes effectively and suggest that CaGe will be useful in the identification of cancer-causing mutations and genes in HGT-based cancer genomics.
In this paper, we present a new cancer genomic tool, CaGe, for the assessment of candidate cancer genes having somatic mutations from HGT-based cancer genomics. CaGe provides users with information on cancer genes, mutations, pathways, and associated annotations through cancer gene annotation, cancer pathway annotation, cancer gene browsing, and cancer pathway browsing functions. It has a capacity to process SIFT or PolyPhen output files as direct input for usual NGS-based cancer genomics flows. Researchers can classify their candidate genes from cancer genome studies into previously known or novel categories of cancer genes and gain insight into the underlying carcinogenic mechanisms through a pathway analysis. We hope that CaGe will be useful for the identification of cancer-causing mutations and genes in HGT-based cancer genomics.