Pathway Analysis of Metabolic Syndrome Using a Genome-Wide Association Study of Korea Associated Resource (KARE) Cohorts
Article information
Abstract
Metabolic syndrome (MetS) is a complex disorder related to insulin resistance, obesity, and inflammation. Genetic and environmental factors also contribute to the development of MetS, and through genome-wide association studies (GWASs), important susceptibility loci have been identified. However, GWASs focus more on individual single-nucleotide polymorphisms (SNPs), explaining only a small portion of genetic heritability. To overcome this limitation, pathway analyses are being applied to GWAS datasets. The aim of this study is to elucidate the biological pathways involved in the pathogenesis of MetS through pathway analysis. Cohort data from the Korea Associated Resource (KARE) was used for analysis, which include 8,842 individuals (age, 52.2 ± 8.9 years; body mass index, 24.6 ± 3.2 kg/m2). A total of 312,121 autosomal SNPs were obtained after quality control. Pathway analysis was conducted using Meta-analysis Gene-Set Enrichment of Variant Associations (MAGENTA) to discover the biological pathways associated with MetS. In the discovery phase, SNPs from chromosome 12, including rs11066280, rs2074356, and rs12229654, were associated with MetS (p < 5 × 10-6), and rs11066280 satisfied the Bonferroni-corrected cutoff (unadjusted p < 1.38 × 10-7, Bonferroni-adjusted p < 0.05). Through pathway analysis, biological pathways, including electron carrier activity, signaling by platelet-derived growth factor (PDGF), the mitogen-activated protein kinase kinase kinase cascade, PDGF binding, peroxisome proliferator-activated receptor (PPAR) signaling, and DNA repair, were associated with MetS. Through pathway analysis of MetS, pathways related with PDGF, mitogen-activated protein kinase, and PPAR signaling, as well as nucleic acid binding, protein secretion, and DNA repair, were identified. Further studies will be needed to clarify the genetic pathogenesis leading to MetS.
Introduction
Metabolic syndrome (MetS) is a complex disorder related to type 2 diabetes mellitus (T2DM) and cardiovascular diseases, and its prevalence is continuously increasing worldwide [1, 2]. Insulin resistance, obesity, and inflammation are major factors leading to MetS; however, the effect of genetic and environmental factors cannot be ignored [3]. Sedentary lifestyle, decreased physical activity, high caloric intake, and westernized food habits are environmental factors leading to obesity and MetS [4, 5]. Parental and maternal obesity in early pregnancy is related to increased risk of childhood obesity, which could later lead to obesity in young adulthood [6, 7]. In addition, family history of obesity, insulin resistance, and T2DM can increase the risk of MetS, implying the importance of genetic contribution. Candidate gene studies in MetS identified genes involved in glucose and insulin signaling, such as insulin receptor substrate 1 (IRS1), peroxisome proliferator-activated receptor γ (PPARG), insulin-like growth factor 1 (IGF1), and genes involved in lipid metabolism, such as adiponectin (ADIPOQ), apolipoprotein A5 (APOA5), and low-density lipoprotein receptor (LDLR) [3, 8, 9, 10]. Through genome-wide association studies (GWASs), a larger number of candidate genes could be further analyzed, and important susceptibility loci were discovered, including fat mass and obesity associated protein (FTO) and the melanocortin 4 receptor gene (MC4R), which were associated with body mass index (BMI) [11, 12]. In another GWAS of MetS, the lipid locus at rs964184 was associated with high-density lipoprotein (HDL)-cholesterol and very low-density lipoprotein-cholesterol [13]. In a meta-analysis in Korea that used the GWAS results from the Korea Associated Resource (KARE) cohort, susceptibility loci in 12q24.11 and 12q24.13 were associated with HDL-cholesterol levels, and genetic factors associated with osteoporosis and metabolic traits, such as T2DM, dyslipidemia, and obesity, could also be identified [14, 15, 16].
GWASs have their strengths in screening susceptible genes associated with complex diseases [17, 18]. However, GWASs focus more on individual single-nucleotide polymorphisms (SNPs) that meet a stringent statistical significance, rather than explaining the interaction of genes, and they can only explain a small portion of genetic heritability [19, 20, 21]. In addition, due to its small effect size, certain SNPs in a GWAS that have been identified to be associated with a disease might not show up in another study of the same disease. This can be seen in two published studies of T2DM and Crohn's disease, which could not find most of the proven susceptibility loci through GWASs and succeeded in achieving moderate significance after replication studies or meta-analysis [22, 23]. To overcome this limitation, pathway-based approaches have been introduced to improve the interpretability of the GWAS.
Pathway-based analysis integrates GWAS data with genes in the selected biological pathways or gene sets from predefined human databases [19, 24]. The strength of pathway analysis is its large effect size and higher power to detect genes that might have been missed through a GWAS [24, 25, 26]. Pathway analysis, such as Meta-analysis Gene-Set Enrichment of Variant Associations (MAGENTA), only requires the SNP p-values and chromosome positions, simplifying the analysis of GWASs [27]. MAGENTA analyzes the statistical power of GWASs through integration of variant association p-values into gene scores, correcting for confounding factors, such as gene size, SNP density, and linkage disequilibrium properties [27]. Through MAGENTA analysis, biological pathways associated with triglyceride, HDL-cholesterol, T2DM, and BMI have been identified [27, 28, 29]. Pathway analysis is a supplementary way to further analyze the results of GWASs. However, there are few studies that have used this approach to identify biological pathways associated with MetS in Asians. The aim of this study was to further elucidate the genomic data of the KARE cohort and to identify the biological pathways related with MetS through a pathway-based approach.
Methods
Subjects
The cohort data from the KARE were used for the analysis. The KARE project, initiated in 2007, is a large cohort study that recruited two population studies from the rural Anseong and urban Ansan cohorts. We analyzed the data of 8,842 individuals (age, 52.2 ± 8.9 years; BMI, 24.6 ± 3.2 kg/m2). Anthropometric measurements, including weight, height, and waist circumference, were measured in all subjects, and BMI was calculated (kg/m2). Systolic and diastolic blood pressures (BP) were examined in all subjects. Fasting plasma glucose and lipid profiles, including serum total cholesterol, HDL-cholesterol, and triglyceride levels, were measured after an overnight fast. Detailed information on the study protocol has been previously described by Cho et al. [16].
MetS was defined according to the modified Third Report of the National Cholesterol Education Program (NCEP-ATPIII) diagnostic criteria, which require the presence of three out of the five following factors: 1) abdominal obesity, defined through waist circumference, using the cut-off values for Asians (≥90 cm in men and ≥80 cm in women), 2) triglycerides ≥ 150 mg/dL or being on lipid-lowering treatment, 3) low HDL-cholesterol (men < 40 mg/dL, women < 50 mg/dL) or being on lipid-lowering treatment, 4) systolic/diastolic BP ≥ 130/85 mm Hg or being on anti-hypertensive treatment, and 5) fasting plasma glucose ≥ 100 mg/dL or previous diagnosis of T2DM or anti-diabetic treatment [30, 31].
Genome-wide association dataset analyses
Genotyping was done using Affymetrix Genome-wide Human SNP Array 5.0 (Affymetrix Inc., Santa Clara, CA, USA). Samples with gender inconsistencies and low call rates (<96%) were excluded.
Quality control (QC) procedures were performed using PLINK version 1.07 [32]. Samples were excluded if there was a high missing call rate (>5%), low minor allele frequency (<0.05), or significant deviation from Hardy-Weinberg equilibrium (p < 1 × 10-6). The total genotyping rate of the remaining individuals was 99.58%. A total of 312,121 autosomal SNPs were obtained after QC, representing 8,842 individuals (4,183 males and 4,659 females). An additive model was used for the analysis. Detailed information on the quality control procedure of the genotypes is described elsewhere by Cho et al. [16].
Pathway-based analysis
Pathway analysis was conducted using MAGENTA (http://broadinstitute.org/mpg/magenta) to discover biological pathways or gene sets associated with MetS. Detailed information on this analysis is described by Segre et al. [27]. Briefly, the steps of MAGENTA analysis were as follows: 1) SNP association p-values and chromosome positions from the GWAS are used as input; 2) each gene located at a predetermined boundary is mapped to a single SNP; 3) based on the regional SNP p-values, gene scores are ranked, and the best SNP p-values are determined; 4) gene scores are corrected for confounding factors, such as gene size and linkage disequilibrium-related properties; and 5) gene set enrichment p-values are determined by analyzing the gene sets enriched with highly ranked gene scores and the selected biological pathway or gene sets [27]. False discovery rate (FDR) was also identified through multiple test correction. Additional information, including 95th and 75th percentile cutoffs and the number of observed and expected genes within each pathway, were also calculated. Since 75th percentile cutoffs have greater power in interpreting complex diseases that are highly polygenic, this cutoff value was used for our interpretation [27, 29, 33].
Results
A total of 8,842 subjects (4,183 men and 4,659 women) were involved in the study. Of them, 3,253 (36.8%) had MetS. Clinical characteristics of subjects with and without MetS are shown in Table 1. Individuals with MetS were older, with higher BMI, systolic BP, triglycerides, and fasting plasma glucose and lower HDL-cholesterol levels compared to subjects without MetS.
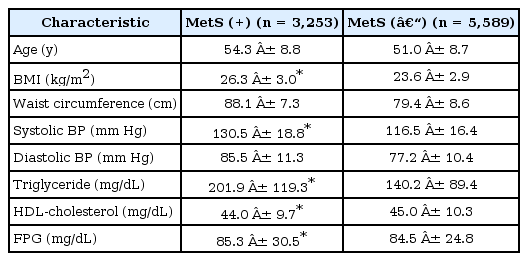
Clinical and biochemical characteristics of subjects with metabolic syndrome and controls
In the discovery set of the GWAS, three SNPs associated with MetS were identified, demonstrated in Table 2. SNPs from chromosome 12, including rs11066280, rs2074356, and rs12229654, had a p < 5 × 10-6. From these SNPs, only rs11066280 satisfied the Bonferroni-corrected cutoff (unadjusted p < 1.38 × 10-7, Bonferroni-adjusted p < 0.05).

Significant SNPs associated with metabolic syndrome in the discovery phase of GWAS
The top 10 significant biological pathways or gene sets associated with MetS at the 75th percentile cutoff are shown in Table 3. The pathways were as follows: electron carrier activity (gene ontology [GO] term), signaling by platelet-derived growth factor (PDGF) (Reactome), mitogen-activated protein kinase kinase kinase (MAPKKK) cascade (GO term), PDGF binding (GO term), nucleic acid binding (protein analysis through evolutionary relationships [PANTHER] molecular function), PPAR signaling (Ingenuity), negative regulation of gene-specific transcription from RNA polymerase II promoter (GO term), non-motor microtubule-binding protein (PANTHER molecular function), protein secretion (GO term), and DNA repair (GO term). At the FDR level, although all pathways were above 0.05, the pathway of electron carrier activity had an FDR value < 0.1, and the pathways of signaling by PDGF and PPAR signaling showed an FDR < 0.2.

Top ten significant GSEA associated with metabolic syndrome at the 75th percentile cutoff
Discussion
In this study, through pathway analysis of MetS, important pathways, including electron carrier activity, signaling by PDGF, MAPKKK cascade, PDGF binding, nucleic acid binding, PPAR signaling, negative regulation of genespecific transcription from RNA polymerase II promoter, non-motor microtubule binding protein, protein secretion, and DNA repair, were identified.
In the previous GWAS using KARE cohorts, rs11066280 and rs2074356 in chromosome 12q24.13, near the chromosome 12 open reading frame, human C12orf51 (C12orf51), and rs12229654 in chromosome 12q24.11, near myosin, light chain 2 (MYL2) were identified to be associated with HDL-cholesterol, hypertension, T2DM, and dyslipidemia [14, 34]. Drinking behavior was also associated with rs11066280 (C12orf51) in Korean men and Han Chinese [34, 35]. In other published GWASs on BMI, important variants on loci near/in FTO, MC4R, and transmembrane protein 18 (TMEM18) were associated with BMI, the latter also having a strong association with BMI in children [36]. Genetic variants in zinc finger protein 259 (ZNF259), lipoprotein lipase (LPL), and APOA5 were also associated with MetS [37]. In the GWAS of European Americans and Finnish cohorts, APOC1 was related with dyslipidemia and central obesity, and the gene cluster region in SNP rs964184, near/in gene APOA1/C3/A4/A5, was associated with MetS [3, 38].
Through MAGENTA analysis, pathways related with electron carrier activity and PDGF signaling and binding, as well as PPAR signaling, were identified as some of the top ranking pathways associated with MetS. Electron carrier activity may be related with electron transport activity in the mitochondria. Abnormal regulation of mitochondrial function is associated with factors, such as reduced electron transport chain, which can lead to insulin resistance and MetS [39, 40]. In obese and diabetic patients, fewer and diminished mitochondrial electron transport enzymes, especially complex I, were observed in the skeletal muscle [41, 42, 43]. Defects in the electron transport chain can impair carbohydrate metabolism, affecting the tricarboxylic acid cycle and limiting ATP activity, which could result in lactic acidosis [44]. In addition, mitochondria respiratory chains are major sites of reactive oxygen species (ROS) production, and excess electrons can increase ROS, stimulating proinflammatory processes and mutagenesis, contributing to mitochondrial dysfunction [40, 45].
Pathways related to PDGF binding and signaling and the MAPKKK cascade were also associated with MetS in this study. The PDGF signaling pathway has been identified to be associated with BMI [29]. PDGF is an important activator of cell proliferation and migration, mediated by the mitogen-activated protein kinase (MAPK) family, and PDGF signaling regulates angiogenesis [46, 47]. In animal studies, PDGF-mediated pathways played a crucial role in healing myocardial infarction, myocardial fibrosis, and defects in the pathway lead to prolongation of inflammation [48, 49]. In a human study, serum PDGF isoform b levels were lower in individuals with MetS, while increased PDGF expression with elevated urinary PDGF-BB was seen in patients with diabetic nephropathy [50, 51]. MAPK pathways are involved in adipogenesis and metabolic homeostasis, and defects in these pathways due to factors, such as oxidative stress, can lead to abnormal adipose regulation, insulin resistance, and obesity [52, 53]. In addition, increased MAPK signaling had a detrimental effect on β-cell function and insulin homeostasis, which could contribute to the development of MetS [54]. The PPAR isotypes PPAR-α, -δ, and -γ play an important role in lipid and glucose metabolism [55]. PPAR-α is expressed in tissues, including skeletal muscle and liver, regulating lipid metabolism and inflammatory processes, whereas PPAR-δ and PPAR-γ are involved in adipocyte differentiation [56]. Genetic variations in PPAR can affect glucose uptake, fasting glucose levels, and BMI [57, 58, 59]. In a GWAS of T2DM in a Finnish population, variants near PPAR-γ were associated with T2DM [60]. In a Korean study, polymorphisms in PPAR-δ were related with BMI and fasting glucose in non-diabetics [61].
Other pathways related with nucleic acids, such as RNA and DNA, as well as pathways of protein secretion were also associated with MetS. MicroRNAs regulate the action and secretion of insulin, as well as lipid metabolism, playing an important role in the pathogenesis of diabetes, obesity, and cancer [62, 63, 64]. Abnormal expression of microRNAs in pancreatic beta-cells affects beta-cell function and insulin secretion [65]. MicroRNA expression is also related to appetite control in the brain; neural signaling in the muscle, pancreas, and liver; and biological processes of lipid metabolism, which are linked to obesity [65, 66]. In addition, microRNA-33 is an important regulator of lipid metabolism, regulating insulin signaling and fatty acid regulation, and may be a therapeutic target for treating MetS [67]. Mitochondrial dysfunction is an important cause, leading to diabetes [68]. DNA damage in mitochondria and vascular cells can have a detrimental effect on mitochondrial function, increasing ROS production and promoting atherosclerosis [68, 69]. Variations in mitochondrial DNA can also lead to MetS, hyperinsulinemia, and T2DM [70, 71, 72]. In addition, insulin also regulates DNA repair, and a chronic hyperglycemic state can damage DNA, contributing to genomic mutation, which can be associated with cancer [73]. Abnormal protein secretion can affect metabolic traits, proven through studies demonstrating increased secretion of fatty acid-binding protein 4 (FABP4), and frizzled-related protein 4 (SFRP4), associated with obesity, insulin resistance, and abnormal insulin sensitivity [74, 75, 76]. Retinol-binding protein 4 (RBP4), expressed in adipocytes and liver, showed a positive correlation with MetS in a Chinese population, associated with insulin resistance and dyslipidemia [77, 78].
The exact association between the pathway of non-motor microtubule binding and MetS can not be explained. However, studies have shown non-motor microtubule binding sites to have an important role in mitosis and to be essential in the embryonic development of Drosophila [79]. More studies will be needed to elucidate the association between non-motor microtubule binding and MetS.
One of the strengths of this study is the usage of a pathway-based approach to further analyze the KARE GWAS datasets. Pathway-based approaches of MetS in Asians are relatively scarce. Therefore, this study might help further elucidate the pathophysiology of MetS. Although the pathways identified in our study did not show an FDR value < 0.05, important pathways related with BMI, lipid and glucose metabolism, including signaling by PDGF and PPAR signaling, had an FDR < 0.2. Other pathway-based approaches will be needed to further validate the identified pathways.
Pathway-based analysis has its strengths in improving the interpretability of the GWAS. However, current pathway analysis tools are limited in finding a well-defined pathway, and their isolated characteristics make it hard to combine them with other analyses [80]. In addition, the limited knowledge base and imprecision of gene annotations restricts their usage and integration with other analysis methods [81]. Therefore, these limitations will need to be improved to generalize this approach and increase its applicability.
In conclusion, through pathway analysis of MetS, significant biological pathways associated with lipid and glucose metabolism could be identified, and these results might contribute to the understanding of MetS.
Acknowledgments
This work was supported by grants from the Korea Centers for Disease Control and Prevention, Republic of Korea (4845-301, 4851-302, 4851-307) and the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2013R1A1A2062702).
Notes
This is 2014 KNIH KARE best paper awarded.