Introduction
Maturity-onset diabetes of the young (MODY) is a monogenic form of diabetes characterized by an early onset, usually before the age of 25 years; an autosomal dominant inheritance; and a defect in pancreatic β-cell function [1]. Depending on the specific gene mutation, the synthesis and secretion of insulin are altered at different stages. MODY is classified according to its affected genes, which include those encoding enzymes, transcription factors, and other types of proteins [1, 2]. The transcription factors associated with MODY act in the nucleus of β-cells and regulate the transcription of INS or other genes encoding enzymes involved in glucose transport and metabolism [1].
Hepatocyte nuclear factor-1β (HNF-1β) is a homeodomain-containing transcription factor that forms a homodimer or heterodimer with structurally related HNF-1α [3]. HNF1B has 9 exons and encodes a 557-amino-acid peptide. Its structure is characterized by a highly conserved DNA-binding domain composed of an atypical POU-specific (POUS) and POU-homeo (POUH) domain, but the molecular properties of HNF-1β have not been studied much. HNF-1 β is known for playing a role in tissue-specific gene expression in organs, including the liver, kidney, and pancreatic islets [4], and is involved in the β-cell transcription factor network [5]. Heterozygous mutations of HNF1B, which encodes HNF-1β, result in maturity-onset diabetes of the young 5 (MODY5), which is characterized by early-onset diabetes and various abnormalities, such as renal cysts, renal impairment, genital malformation, or hepatobiliary involvement [6, 7, 8, 9]. Patients with mutations in HNF1B have impaired insulin secretory responses to glucose and insulin secretagogues [7, 10, 11] and show progressive loss in basal insulin secretion.
Mutation in HNF1B was first described by Horikawa et al. in 1997 [12]. Different mutation types, including missense, nonsense, and frameshift mutations, have been found in different domains [13, 14, 15]. Recently, mutations in exon 2 and in the DNA-binding domain have been reported [16, 17, 18, 19, 20]. Barbacci et al. [21] characterized eight naturally occurring mutations in different domains. Truncated mutations showed defective nuclear localization and weak dominant-negative activity, whereas a frameshift mutation within the QSP-rich domain had partially reduced transcriptional activity. Missense mutations in POUS and POUH exhibited severe decreases in transcription. A certain mutation showed a gain-of-function phenotype [22, 23]. In vitro studies suggested that clinical phenotypes may be related to loss of function and/or dominant-negative mechanisms [8, 24].
In this study, we have identified a family with MODY5 harboring a heterozygous P159L HNF1B mutation. We evaluated the functional consequences of this HNF1B mutation on glucose metabolism.
Methods
Sequencing of HNF1B
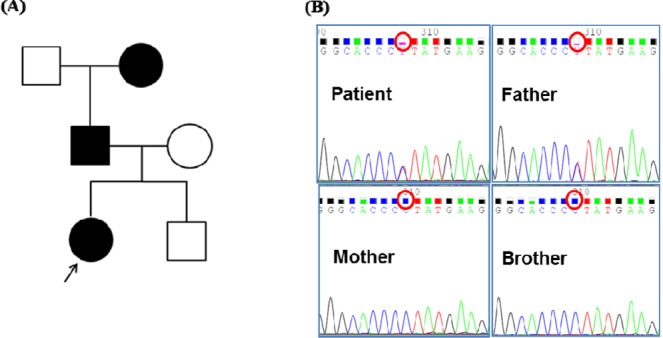
The HNF1B of the patient was sequenced by Sanger method in peripheral blood DNA. Her father, mother, and younger brother were also screened by the same method. Written informed consent for the genetic study was obtained from the patient and her family members before sequencing.
Wild-type and mutant plasmid constructs
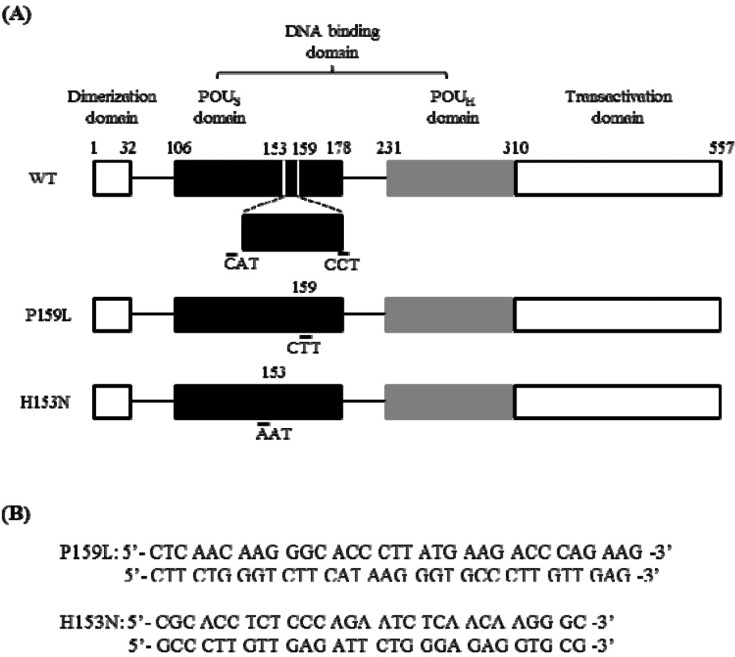
Human wild-type HNF1B, cloned in pCMV6b, was provided by Dr. J. Takeda, Gifu University, Japan. A point mutation was generated by PCR-based site-directed mutagenesis using the QuickChange Mutagenesis Kit (Stratagene, La Jolla, CA, USA). The promoter of human glucose transporter type 2 (GLUT2) (-1296 to +312), cloned in the pGL3-Basic vector (Promega, Madison, WI, USA), was provided by Dr. M.S. Lee, Sungkyunkwan University, Korea. The sequences of all genes for the experiment were confirmed by direct nucleotide sequencing.
Cell culture
COS-7 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA), supplemented with 100 U/mL of penicillin and 100 µg/mL of streptomycin, before transient transfection. MIN6 cells were cultured in RPMI 1640 with 11.1 mM D-glucose supplemented with 10% fetal bovine serum (Invitrogen) and the same antibiotics as for COS-7. Cells were cultured at 37℃ in 5% CO2:95% air.
Transient transfection
COS-7 cells were subcultured in 12-well plates the day before transfection; 0.2 µg of pGL3-GLUT2 promoter, 0.02 µg of HNF1B expression plasmids, and 0.05 µg of pCMV-β-galactosidase were combined with LipofectAMINE PLUS agent (Invitrogen) according to the manufacturer's protocol. The cells, in 400 µL of serum-free DMEM, were treated with the complex for 3 h, and the medium was changed to DMEM with 10% fetal bovine serum. MIN6 cells were subcultured in 6-well plates the day before transfection. As it is difficult to transfect genes into MIN6 cells, the transfection efficiency was evaluated with the green fluorescent protein gene (GFP) first. After the efficiency was confirmed, 0.5 µg of expression vector for HNF1B per well was administered. Serum-containing medium was added up to the normal volume after 3 h of incubation and replaced with fresh complete RPMI 1640 after 8 h. MIN6 cells were harvested for RNA extraction and protein quantitation after 30 h after the start of transfection.
Luciferase reporter assays
Twenty-four hours after the start of transfection, Cos7 cells were lysed with reporter lysis buffer and harvested, and the transcriptional activity was measured using the Luciferase assay system (Promega) and Lumet LB9507 (Berthold, Bad Wildbad, Germany) according to the manufacturer's instructions. To normalize transfection efficiency, β-galactosidase activity was measured. Ten microliters of cell lysate was mixed with 3 µL of 100 × MgCl2 (0.1 M MgCl2 and 4.5 M β-mercaptoethanol) and 66 µL 1 × o-nitrophenyl-β-D-galactoside. The activity of β-galactosidase was measured by VersaMax spectrophotometry at 420 nm (Molecular devices, Sunnyvale, CA, USA).
Electrophoretic mobility shift assay
Double-stranded oligonucleotides composed of the following sequences were used for human GLUT2 promoter probes: 5'-AAG ACC TCA GTA AAG ATT AAC CAT CAT TA-3'; 1 µg of up and down sequences of single-stranded oligomer were hybridized within PE buffer (20 mM Tris, 10 mM MgCl2, 50 nM NaCl, 1 mM dithiothreitol [DTT]) by incubating them at 72℃ for 10 min and then at room temperature for 15 min. The probe was labeled with [α-32P] dATP by Klenow DNA polymerase (Ambion, Austin, TX, USA), and the labeled probe (25,000 cpm) was incubated with nuclear extracts of COS-7 cells that were transfected with pcDNA, wild-type HNF1B, and H153N and P159L HNF1B in 10 mM HEPES (pH 7.9, Sigma-Aldrich, St. Louis, MO, USA) containing 50 mM KCl, 0.1 mM EDTA, 0.25 mM DTT, 0.1 mg/mL poly(dIdC), 0.01% Nonidet P-40, and 10% glycerol at room temperature. DNA-protein complexes were separated on 6% polyacrylamide gels and detected by autoradiography.
RNA isolation and real-time PCR
Mutant or wild-type HNF1B was overexpressed in the mouse pancreatic β-cell line MIN6. Total RNA was isolated using TRIzol (Invitrogen) according to the manufacturer's instructions. To prepare cDNA, 6 µL of reaction buffer (Invitrogen), 3 µL of 100 mM DTT, 1.5 µL of 10 mM dNTP, 0.6 µL of random primer, 0.3 µL of RNase inhibitor, 1.0 µL of RTase, and 1 µg of RNA were mixed, and RNase-free water was added up to 30 µL. The mixture was incubated 37℃ for 1 h and 72℃ for 10 min using a PCR system. Expression levels of genes were determined by using SYBR Master Mix (Takara, Otsu, Japan) and an AB 7500 Real-time PCR system (Applied Biosystems, Foster City, CA, USA). The primer sequences for the PCR were as follows: 5'-GGC TAA TTT CAG GAC TGG TT-3' and 5'-TTT CTT TGC CCT GAC TTC CT-3' for GLUT2 and 5'-CTC GGC TCA CCG TTT CCT T-3' and 5'-CGC GCT TGC TAA TAG TGC AG-3' for INS. Samples were prepared repetitively, and each sample was analyzed in duplicate. Expression levels of each gene were measured relative to 18S and normalized to the expression levels in total RNA with the ΔΔCT method.
Western blot analysis
Cells were lysed in a buffer containing 20 mM Tris-HCl, pH 7.4, 5 mM Na4P2O7, 100 nM NaF, 2 mM Na3VO4, 1% NP-40, and protease inhibitor (1 µg/µL of aprotinin, 1 µg/µL of leupeptin, and 1 mM of PMSF; Sigma-Aldrich). After determination of protein concentrations, equal amounts of cell lysates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis. Separated proteins were transferred onto a nitrocellulose membrane (Whatman, Dassel, Germany). The membrane was blocked with 5% skim milk in Tris-buffered saline-Tween-20 (TBS-T) for 1 h at room temperature and incubated with rabbit polyclonal anti-HNF-1β antibody (Santa Cruz Biotech, San Diego, CA, USA) at 4℃. After 8 h, the membranes were probed with secondary antibody, and bands were visualized by enhanced chemi-luminescence (Pierce, Rockford, IL, USA).
Statistical analysis
Data were expressed as mean ± standard error (SE). The non-parametric t-test and one-way ANOVA were used to compare the mean values between the groups with GraphPad Prism version 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). p < 0.05 was considered statistically significant. All individual experiments were performed at least in duplicate and were repeated five times on separate occasions.
Results
A 27-year-old woman with inadequately controlled diabetes mellitus was admitted for glycemic control. She was diagnosed as having diabetes mellitus at an age of 14 (fasting plasma glucose 137 mg/dL, postprandial 2-h glucose 239 mg/dL), with normal body mass index (21.9 kg/m2) and 24-h urine C-peptide level (83.3 µg/day; reference range, 50 to 100µg/day). Although autoantibodies for pancreatic β-cell were not detected, she required insulin therapy for glycemic control. Her serum C-peptide level decreased from 1.8 ng/mL to 0.9 ng/mL during the past year. The total daily insulin requirement was 0.68 IU/kg. The patient's father (diagnosed in his early 30s) and grandmother (the age of diagnosis was unknown) also had diabetes. She did not have urogenital anomalies.
Sequencing of HNF1B revealed a heterozygous P159L point mutation (CCT to CTT in codon 159) in the patient. Among the three other family members, her father was found to have the identical mutation. The pedigree of diabetes mellitus and the result of Sanger sequencing are shown in Fig. 1. This nonsynonymous variant co-segregated according to the affected state of diabetes within the family.
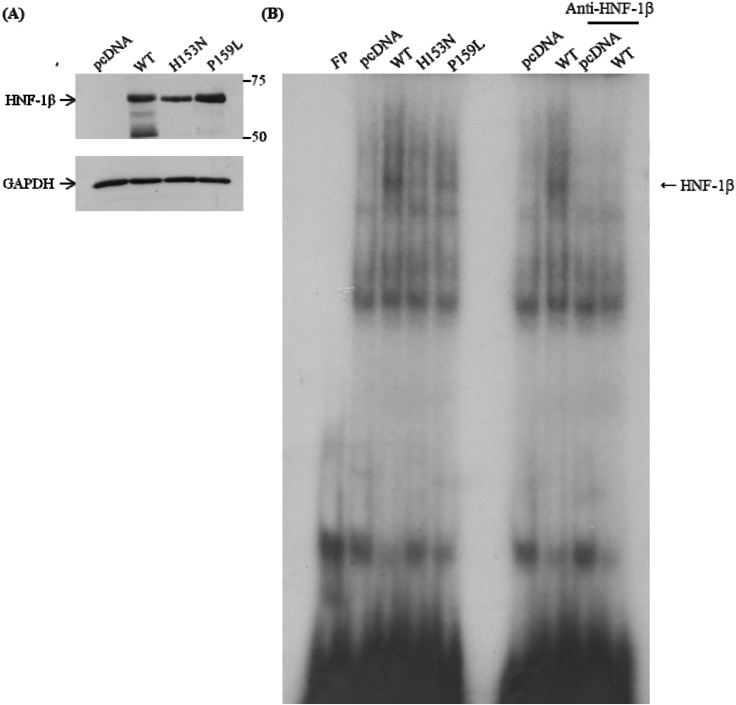
Two point mutations, P159L and H153N (for comparison as functionally proven mutations; i.e., positive control), were generated by in vitro mutagenesis (Fig. 2). Western blot analysis confirmed that the mutant protein was apparently expressed in the transfected COS-7 cells (Fig. 3A). The region, including nucleotide -1030 of human GLUT2 promoter, which is known to be the cis-element for HNF-1β binding [24], was inserted into the luciferase vector (Fig. 3B). Luciferase activity of the human GLUT2 promoter was increased with overexpression of wild-type HNF-1β compared to the control (p < 0.05 vs. control) (Fig. 3C). When P159L HNF-1β and H153N HNF-1β were expressed, a significant reduction of transcription activity was observed (p < 0.05 vs. wild-type, respectively). In the electrophoretic mobility shift assay (EMSA), the proteins of the expected sizes were translated (Fig. 4A). The EMSA showed that wild-type HNF-1β protein bound well to the GLUT2 promoter, but the mutant proteins had decreased binding activity (Fig. 4B).
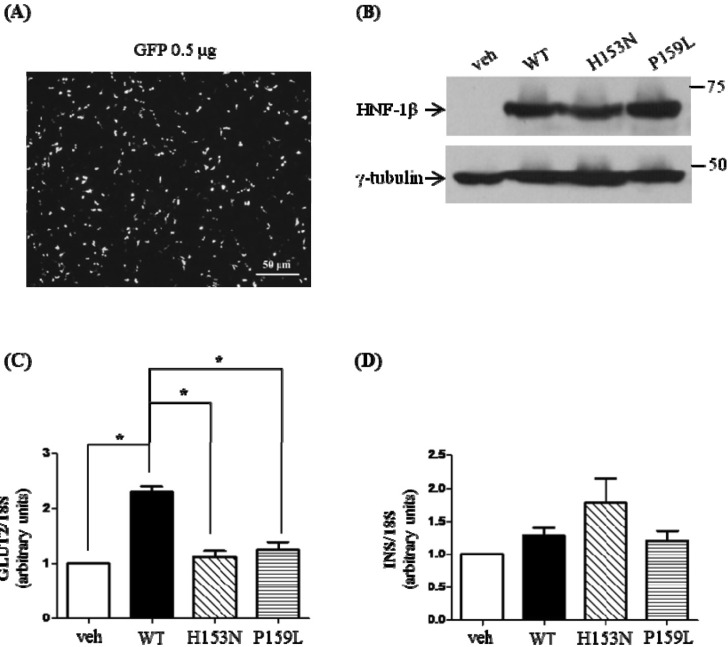
The mRNA levels of GLUT2 and INS in MIN6 cells after HNF1B transfection were also investigated. The transfection into MIN6 was confirmed with an efficiency of 60% to 65% (Fig. 5A). Western blot analysis showed that the protein expression was well preserved after transfection (Fig. 5B). The quantitated mRNA levels were normalized to that of 18S. When the wild-type HNF-1β was overexpressed, the relative mRNA level of GLUT2 was increased by 2.3-fold (p < 0.05 vs. control) (Fig. 5C). Compared to the wild-type HNF-1β, the relative mRNA levels of GLUT2 for the H153N and P159L mutants were significantly decreased (p < 0.05 vs. wild-type, respectively). There was no significant difference in the mRNA expression level of INS among the four groups (Fig. 5D).
Discussion
In this study, we identified a MODY5 family with a nonsynonymous mutation in the HNF1B gene. The patient was suspected to have MODY, especially MODY5, because of the lack of response to sulfonylureas, early (within 3 years) requirement of exogenous insulin, and features of insulin resistance [25]. Although the patient did not have specific clinical features of HNF1B mutation, such as cystic kidney disease [6], she had fulfilled the clinical diagnostic criteria of MODY. The clinical features of MODY5 vary as either diabetes or urinary tract malformation, or both could be the presenting symptoms [14, 20, 26]. According to a systematic review performed by Chen et al. [27], renal structural anomalies are relatively less frequent in missense mutations than in other types of mutation. On the contrary, the percentage of diabetes mellitus and the frequency of insulin treatment tended to be higher.
The mutation of P159L was heterozygous c.476C>T in exon 2 of HNF1B, which is in the DNA-binding domain. Chen et al. [27] found that mutations in HNF1B were more likely to be located in the DNA-binding domain, and the hotspots were in exons 2 and 4. Yamagata [28] reviewed that N228K is a relatively common polymorphism in Pima Indians, and the A241T, G492S, and S465R mutations might be rare polymorphisms. In HNF1B-deficient mouse embryos, a dorsal bud of the pancreas is formed transiently and becomes absent, whereas the ventral bud is not affected [29]. In two 8-week human fetuses with HNF1B mutations, severe pancreas hypoplasia was observed [30]. With these consistent findings, the abnormality in the morphogenesis of the pancreas is thought to be one of the causes of diabetes in MODY5 patients.
Another suggested molecular mechanism is related to altered GLUT2 expression. Wild-type HNF-1β is known to bind to the cis-element of GLUT2 and efficiently increase its transcription [24]. In this study, overexpression of wild-type HNF-1β resulted in an increase in transcriptional activity of the GLUT2 promoter, whereas overexpression of the P159L mutant resulted in decreased GLUT2 expression. Previous studies showed similar results in various types of cells, such as COS-7, MIN6, and HepG2 [20, 24]. GLUT2 is a low-affinity, high-capacity transporter expressed mainly in hepatocytes, renal proximal tubule cells, and pancreatic β-cells [31, 32]. GLUT2 plays a role in β-cell insulin secretion, as it senses external glucose and transports it into pancreatic β-cells [33]. In vivo studies showed that GLUT2-deficient mice were hyperglycemic, resulting in diabetes with impaired glucose sensing by pancreatic β-cells [34], and GLUT2 was undetectable on β-cells of the two HNF1B mutant fetuses [29]. So, decreased expression of GLUT2 in β-cells, combined with a gradual reduction in insulin secretion, could be involved in the pathogenesis of diabetes mellitus in MODY5 patients. In addition, GLUT2 may also affect β-cell differentiation in the development of the pancreas [34, 35].
EMSA was done to examine whether the reduced transcription activity was due to the decreased DNA-binding capacity. Both mutant HNF-1β proteins had decreased DNA-binding activity compared to the wild-type. It was previously reported that the H153N HNF-1β mutation had diminished DNA-binding activity [20]. Since both P159L and H153N mutants have intact dimerization domains, it is possible that they could dimerize with wild-type HNF-1β [9]. However, the mutant/wild-type dimer seems to have a defect in DNA binding and limited ability to increase the transcription of GLUT2. The mRNA expression of GLUT2 was increased with wild-type HNF-1β but significantly suppressed, almost to the level observed in the empty control vector, in both mutants. The findings of the real-time PCR analysis of GLUT2 were consistent with the reporter gene assay and DNA binding assay.
On the other hand, there has been limited knowledge regarding the mRNA expression of INS with mutant HNF-1β. The mRNA levels of INS were not statistically different between the wild-type- and mutant-expressing MIN6 cells. In one study with HNF-1α, neither the wild-type nor the mutant activated GLUT2 or the insulin promoter in MIN6 cells [36]. The authors concluded that endogenous HNF-1α, GLUT2, and insulin proteins in MIN6 cells could interfere in the transfected reporter assay. The mRNA expression of INS is also considered to be regulated by other transcription factors, and it might have been compensated for by complex mechanisms against the mutation of HNF1B. Overall, the impaired insulin secretion in MODY5 patients might be explained by pancreatic hypoplasia or β-cell dysgenesis rather than decreased mRNA expression of INS. As described above, decreased mRNA expression of GLUT2 might also result in the inhibition of insulin secretion.
This study has some limitations. First, we could not sequence HNF1B of the patient's grandmother, who was also a patient with diabetes. If she had the same P159L mutation of HNF1B, it would have revealed an autosomal dominant inheritance more clearly. Second, alterations in insulin secretion with the mutation in the pancreatic β-cell line were not evaluated. Further studies of functional changes in the P159L mutation are needed.
In conclusion, we have identified a P159L mutation of HNF1B in a family with MODY5 and have characterized the functional consequences of this nonsynonymous mutation in the pathogenesis of diabetes. This mutation revealed decreased DNA-binding activity and decreased transcriptional activity of GLUT2. The mRNA expression of GLUT2 in pancreatic β-cell lines was also significantly decreased with this mutation, whereas that of INS was not altered. Impaired insulin secretion in the patient with the P159L mutation in HNF1B might be associated with the alteration of GLUT2-related signaling pathways.